
Gangliocytomas are rare indolent CNS tumours (WHO grade 1), primarily encountered in children and frequently discovered as the cause of epilepsy. They are considered one of the long-term epilepsy-associated tumours (LEATs).
They differ from gangliogliomas by the absence of neoplastic glial cells, although both tumours are defined by the presence of displaced ganglion cells (large mature neurones that show cytological or architectural abnormalities).
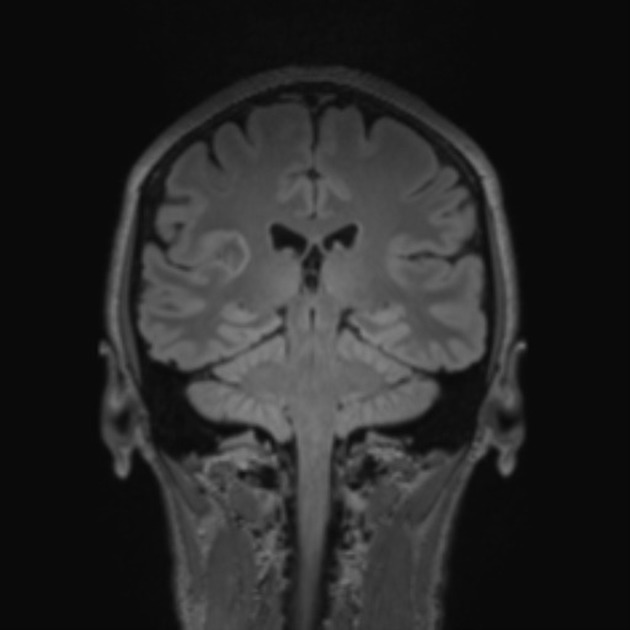
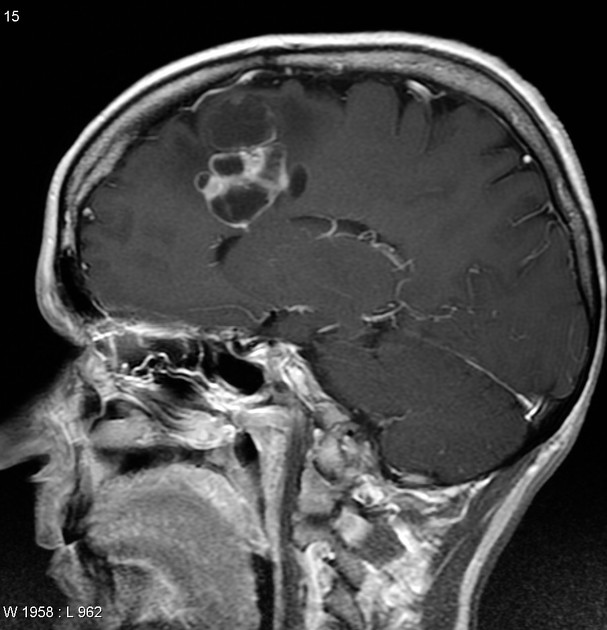
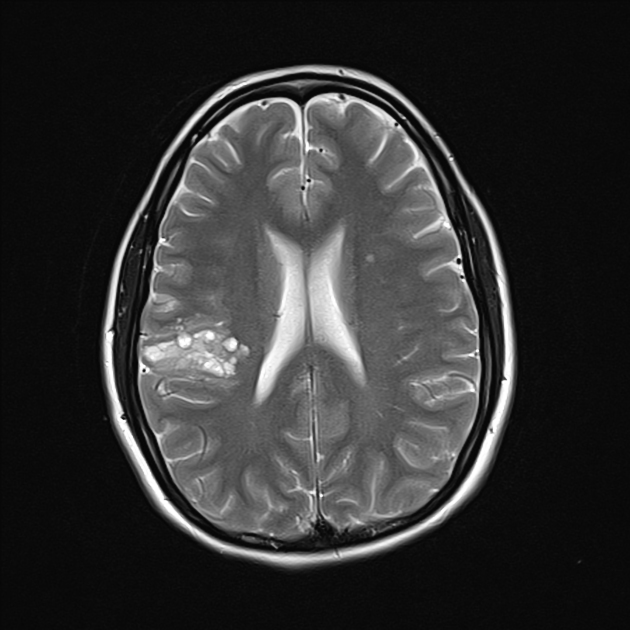
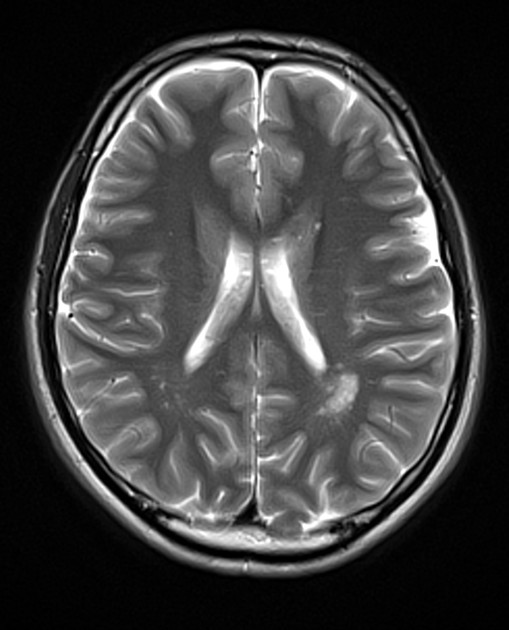
On imaging, these tumours are usually characterised by cortical solid lesions with little associated mass effect and minimal or no surrounding vasogenic oedema. Calcification and cyst formation can occur, and contrast enhancement is generally present.
On this page:
Terminology
Gangliocytomas should not be confused with dysplastic cerebellar gangliocytoma of the cerebellum, better known as Lhermitte-Duclos disease.
Epidemiology
Gangliocytomas account for 0.1-0.5% of all brain tumours but 1-3% of tumours in epilepsy series 5. They tend to be diagnosed in children and young adults, with a peak age between 10 and 30 years of age 6.
Clinical presentation
No clinical differences between gangliogliomas and gangliocytomas are recognised. Tumours in the cerebral cortex, particularly the temporal lobe, present most commonly with epilepsy which is often longstanding at the time of diagnosis 4,6.
Pathology
Gangliocytomas may arise anywhere within the neuroaxis, however, over 80% are found in the temporal lobes 4-6.
Microscopic appearance
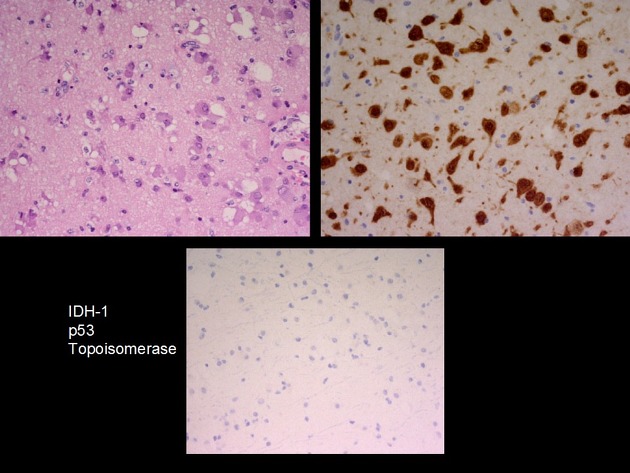
The tumour is composed of abnormal large mature neurones, usually with a multipolar morphology 5. Some neurones are binucleated and cytoplasmic vacuolization or ballooning is seen 5. Microcalcification can also be seen 5.
The key feature in distinguishing gangliocytomas from gangliogliomas is identifying a lack of neoplastic glial cells. Similarly, ensuring no small round blue cell component or neuroblasts are seen is important to exclude more aggressive tumours (such as medulloblastoma and neuroblastoma) which can have similar appearing ganglion cells 5.
Immunophenotype
The pattern of immunostaining is consistent with neuronal components 5,6.
synaptophysin: positive
neurofilament: positive
chromogranin-A: positive
MAP2: positive
NFP: variable
GFAP: negative
Radiographic features
Appearances of gangliocytomas are indistinguishable from gangliogliomas 5,6. They tend to occur in the cortex, most often in the temporal lobes. Because of cortical location and slow growth, calvarial remodelling may be seen in some cases 6.
CT
Gangliocytomas typically appears hyperattenuating on non-contrast imaging. They usually only have little associated mass effect and minimal or no surrounding vasogenic oedema. Calcification and cyst formation can occur 1.
MRI
Gangliocytomas often have cystic and solid components and may demonstrate calcifications 6.
T1: solid components typically hypointense
T2: solid components are typically mildly hypointense 2; cystic areas are hyperintense; calcification if present can be hypointense
T1 C+ (Gd): solid components enhance
T2*/SWI: signal loss due to calcification
Treatment and prognosis
These tumours tend to grow slowly and do not undergo anaplastic change, unlike gangliogliomas that can (rarely) have higher grade glial components. Resection is curative with a 7.5 year progression-free survival rate of 94% 6.
Differential diagnosis
The differential diagnosis is primarily that of other long-term epilepsy-associated tumours (LEATs) including:
-
can be indistinguishable on imaging
more common than gangliocytoma
-
polymorphous low grade neuroepithelial tumour of the young (PLNTY)
can be indistinguishable on imaging
less common
-
can be indistinguishable on imaging
rare
-
pleomorphic xanthoastrocytoma (PXA)
contrast enhancement prominent
dural tail sign is sometimes seen
-
dysembryoplastic neuroepithelial tumours (DNET)
contrast enhancement uncommon
-
usually cerebellar or optic pathway (esp in NF1)
when suptratentorial often near the ventricles
-
calcifications common
less commonly in the temporal lobes


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.