
Papillary glioneuronal tumors are rare well circumscribed complex solid cystic supratentorial lesion with an indolent clinical course. They are often considered part of the heterogeneous group of tumors known as long-term epilepsy-associated tumors (LEATs).
On this page:
Epidemiology
These tumors typically are diagnosed in younger patients (median age at diagnosis 23 years) but are reported essentially in all ages 4. No sex predilection has been identified 4.
Clinical presentation
Clinical presentation is non-specific, primarily comprising focal seizures/epilepsy and/or headaches. Focal neurological deficits may also be present.
Pathology
Papillary glioneuronal tumors were first recognized in the 2007 edition of the WHO classification of CNS tumors and are considered WHO grade 1 tumors 6.
The majority of these tumors are in the cerebral hemispheres, with occasional intraventricular tumors described 4.
Macroscopic appearance
These tumors usually demonstrate a mixture of solid and cystic components, sometimes with calcification 4.
Microscopic appearance
Papillary glioneuronal tumors contain both glial (astrocytic) and neural elements. Characteristic microscopic features are hyalinized vascular pseudopapillary architecture with a pseudostratified layer of cuboidal glial cells and focal collections of neurocytes and ganglion cells.
Immunophenotype
Immunohistochemistry of the cuboidal glial cells reflects their glial appearance 4:
In contrast, the neuronal components show a separate immunophenotype 4:
synaptophysin: positive
neuron-specific enolase: positive
class III beta-tubulin: positive
These tumors demonstrate a low Ki-67 proliferation index/MIB-1 index ~1-2% 1-4. Rarely, tumors with higher mitotic index are encountered and some have more aggressive behavior 4.
Genetics
Papillary glioneuronal tumors have a unique fusion oncogene SLC44A1-PRKCA resulting from a translocation t(9; 17)(q31; q24) 5.
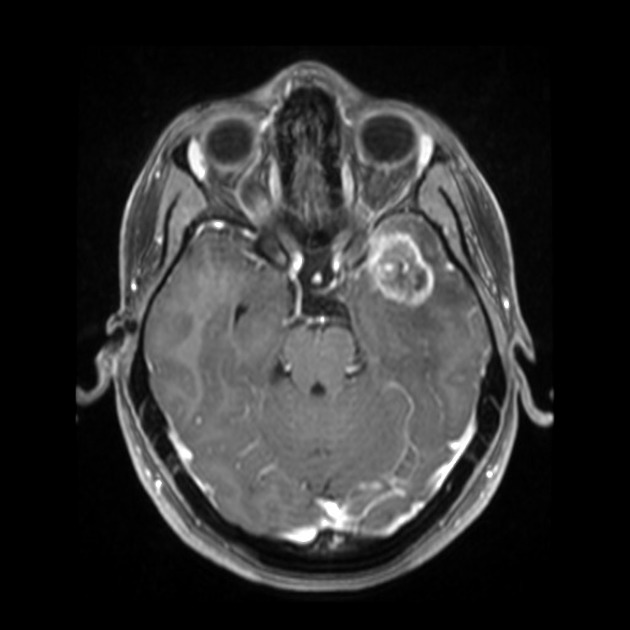
Radiographic features
Features on imaging are very similar to ganglioglioma. They are supratentorial solid-cystic or cystic with solid nodule lesions showing intense but heterogeneous enhancement. Calcification is a frequent finding 1-4. Hemorrhage is seen in ~10% of cases and is sometimes very pronounced 4.
MRI
T1: isointense to hypointense
T2: inhomogeneously hyperintense
-
FLAIR:
inhomogeneously hyperintense nodule
cystic components may suppress
T1 C+: avid but heterogeneous enhancement of the solid nodule
Treatment and prognosis
Papillary glioneuronal tumors are indolent and surgical resection is usually sufficient to effect a cure. In cases where proliferation is high (e.g. Ki-67 >5%) then local recurrence has been described 4.
Differential diagnosis
The differential diagnosis is primarily of other parenchymal tumors with mixed solid and cystic components, including:
-
gangliogliomas and gangliocytomas
can be indistinguishable on imaging
less common
-
polymorphous low grade neuroepithelial tumor of the young (PLNTY)
can be indistinguishable on imaging
less common
-
pleomorphic xanthoastrocytoma (PXA)
contrast enhancement prominent
dural tail sign is sometimes seen
-
dysembryoplastic neuroepithelial tumors (DNET)
contrast enhancement uncommon
-
usually cerebellar or optic pathway (especially in neurofibromatosis type 1)
when supratentorial, often near the ventricles
-
rosette-forming glioneuronal tumors
relevant particularly if midline/posterior fossa


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.