
Prion diseases, also known as transmissible spongiform encephalopathies, are caused by the accumulation of dysmorphic proteins named prions, and are characterised by progressive neurological decline and eventual death.
In humans, prion diseases include:
Creutzfeldt-Jakob disease (sporadic, variant, familial and iatrogenic)
In addition to the above, other neurodegenerative diseases have been postulated to have a prionopathic aspect to their pathophysiology, for example, iatrogenic cerebral amyloid angiopathy, iatrogenic Alzheimer disease, and certain alpha-synucleinopathies 5-7.
In non-human animals, prion diseases include 3,4:
bovine spongiform encephalopathy ("mad cow disease): in cows, evidence of transmission to humans (known as variant Creutzfeldt-Jakob disease)
scrapie: in sheep, no evidence of transmission to humans
chronic wasting disease: in deer and elk, no evidence of transmission to humans
transmissible mink encephalopathy: in minks, no evidence of transmission to humans
feline spongiform encephalopathy: in felines, no evidence of transmission to humans
exotic ungulate spongiform encephalopathy: in ungulates (e.g. antelopes), no evidence of of transmission to humans
camel spongiform encephalopathy ("mad camel disease"): in camels, no evidence of transmission to humans
On this page:
Pathology
Prion disease are all mediated by prions, a term derived from 'protein' and 'infection. Prions are abnormally folded self-catalysing endogenous proteins which accumulate within the nervous system.
Diagnosis has been problematic as imaging findings are variable and CSF protein analysis (14-3-3 protein) insensitive. Brain biopsy is accurate but is fraught with difficulties as equipment needs to be discarded (prions have been found to survive standard autoclaving procedures) and staff precautions are difficult to implement.
Radiographic features
MRI
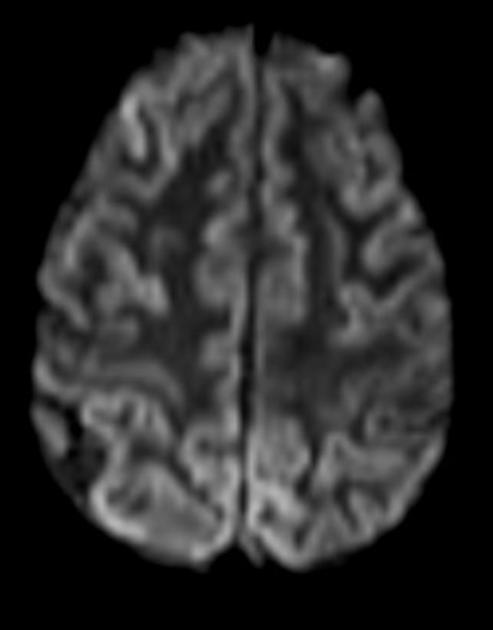
Generally, changes on DWI are the earliest imaging manifestation, prior to FLAIR and cerebral volume changes. Typically, cortical and deep grey matter (caudate, thalamus and putamen) demonstrates increased signal, not confined to vascular territories, although the exact pattern of involvement depends on the specific prion disease.
Treatment and prognosis
There is no curative treatment available for any prion disease and they are all universally fatal.
History and etymology
The term spongiform is derived from the histological appearance of cerebral tissue characterised by neuronal degeneration with peculiar vacuolation reminiscent of a sponge.
Although Creutzfeldt-Jakob disease had been clinically described in 1921, it was not until 1959 when a similarity between kuru (only described in 1957) and scrapie (a similar disease affecting sheep) was recognised by veterinary pathologist W J Hadlow, that prion disease was recognised as a group of related pathologies 2.



 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}