
Lymphoid interstitial pneumonia (LIP), also known as lymphocytic interstitial pneumonitis, is a benign lymphoproliferative disorder characterised by lymphocyte-predominant infiltration of the lungs. It is classified as a subtype of interstitial lung disease. It also falls under the umbrella of non-lymphomatous pulmonary lymphoid disorders.
On this page:
Epidemiology
Lymphoid interstitial pneumonia can occur at any age. However, most of the patients are adults with a mean age of 52-56 years. If a child presents with lymphoid interstitial pneumonia, this can be indicative of AIDS.
Females are twice as likely to be affected, most likely because of its association with autoimmune diseases such as Sjögren syndrome, which is by far more common in women 8.
Associations
-
considered the most common lung pathology in these patients 14
can occur in up to 25% of those with lymphoid interstitial pneumonia 6
AIDS: particularly if it occurs in the young 9
Clinical presentation
The main clinical symptoms are a gradual onset of dyspnoea and cough with approximately 6 months duration. Less frequently, patients may have systemic symptoms such as fever, night sweats, arthralgia, and weight loss. If the disease progresses to end-stage respiratory failure, cyanosis, and clubbing may develop. Hypertrophy of the salivary glands may be seen in 20% of patients 11.
Pathology
It is considered a benign lymphoproliferative disorder characterised histologically by diffuse interstitial and alveolar infiltration with polyclonal lymphocytes and plasma cells.
Markers
In about 80% of patients polyclonal or IgM monoclonal gammopathy is found 8.
Radiographic features
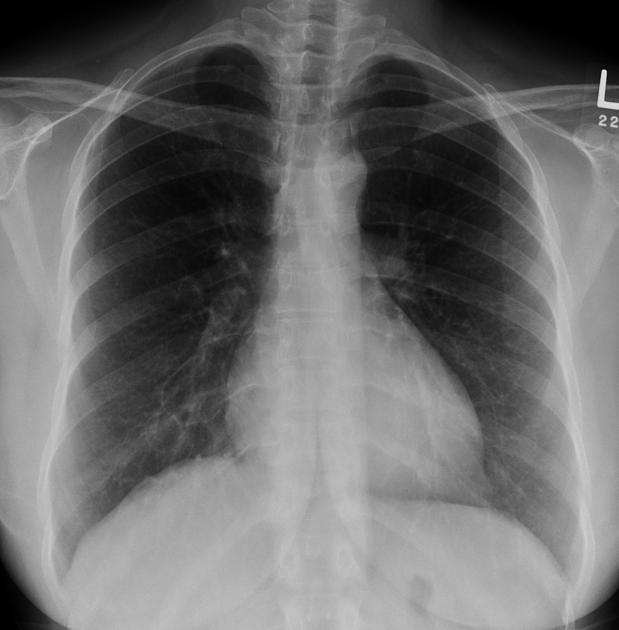
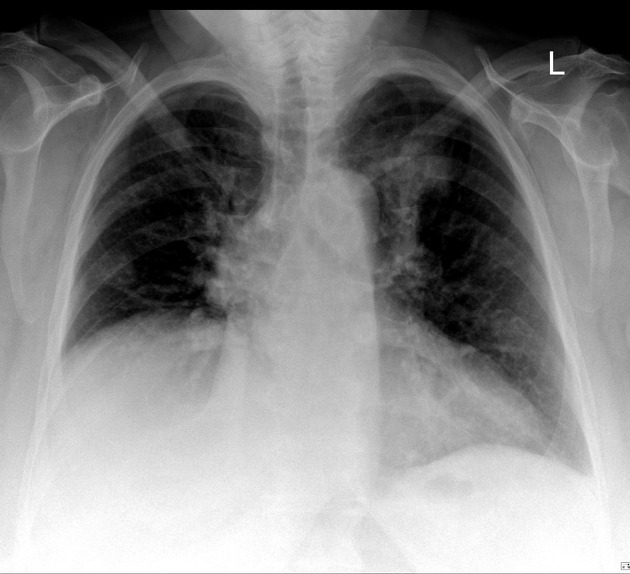
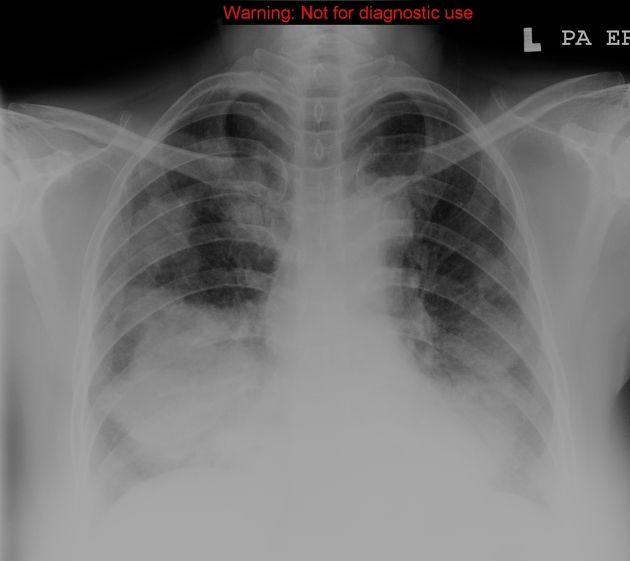
Plain radiograph
Features can be non-specific, but may include:
lower-zone predominant bilateral reticular opacification
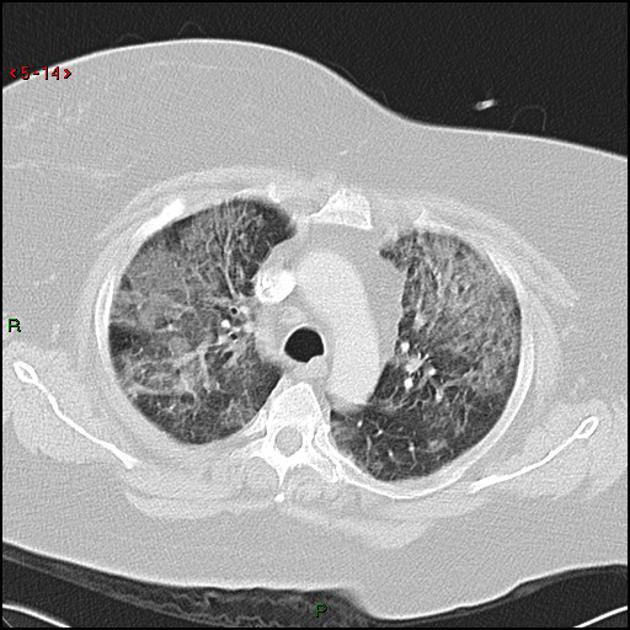
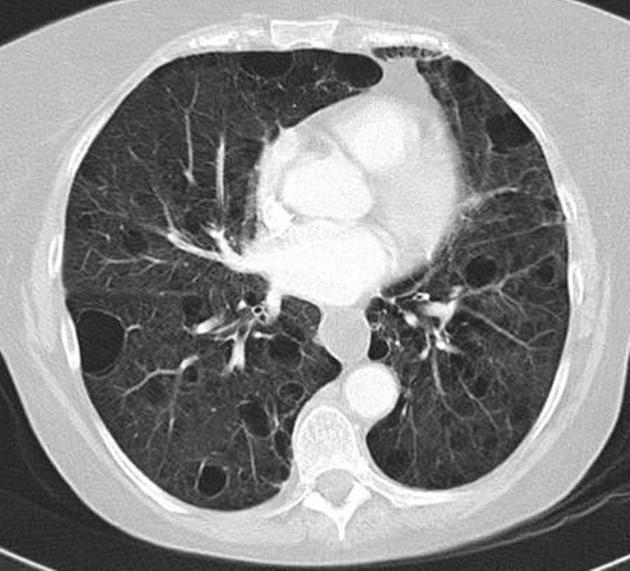
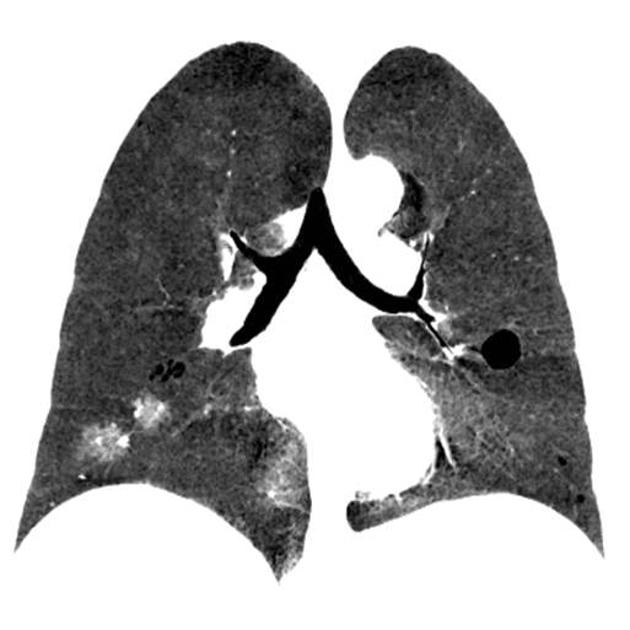
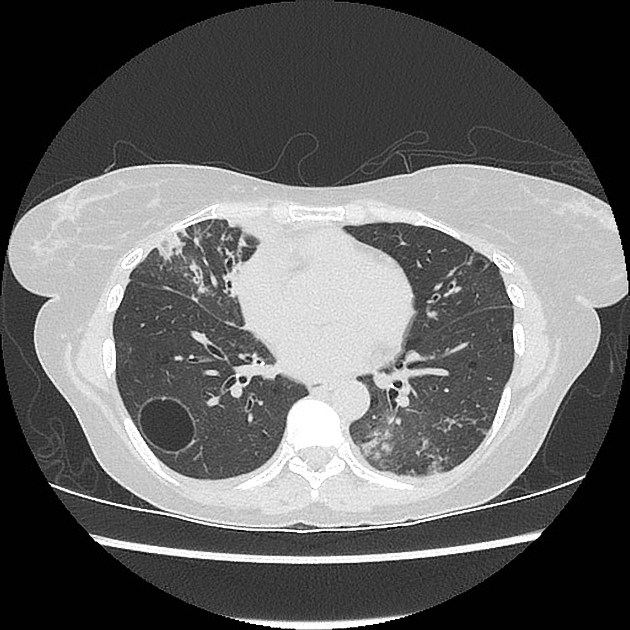
CT
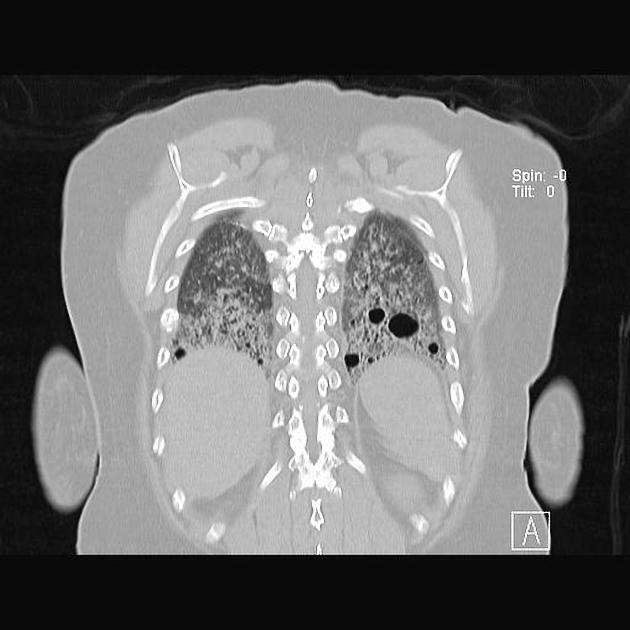
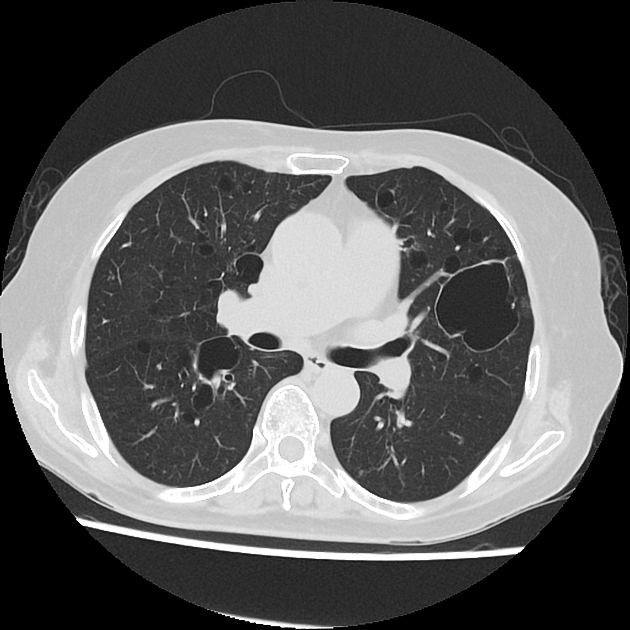
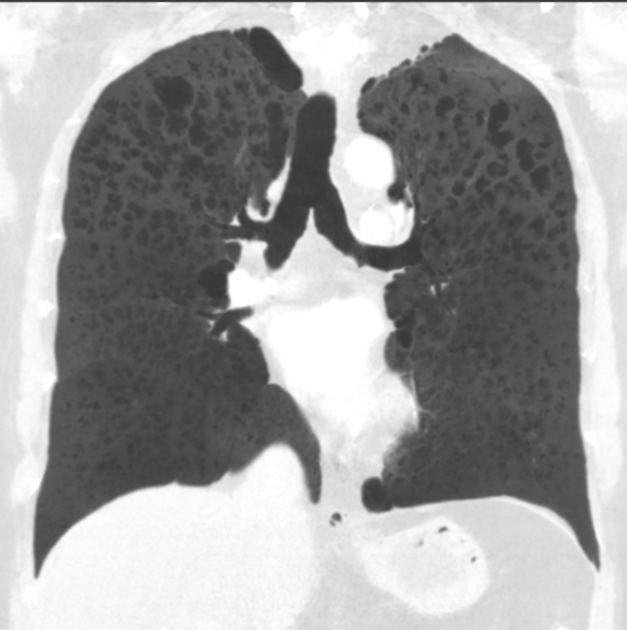
The following features may be seen with lymphoid interstitial pneumonia on HRCT, but the findings are not exclusive to its diagnosis:
features tend to be diffuse with mid to lower lobe predominance
interstitial thickening along lymph channels 2
small but variable-sized pulmonary nodules (can be centrilobular or subpleural, and are often ill-defined)
Scattered thin-walled perivascular cysts are commonly seen in Sjogren’s syndrome however these are most likely due to multifocal lung destruction by macrophage metalloproteinases rather than LIP. Macrophages are attracted to sites of protein deposition and the thin-walled cysts may contain internal strands, identical to those seen in light chain deposition disease and other lymphoplasmacytic disorders 16, 17.
Unfortunately current classifications still include cysts as a feature of 'radiological' LIP in contrast to pathological LIP which does not include cysts.
Treatment and prognosis
The natural history is variable, from near-complete resolution to progressive disease. More than 30% of patients will develop end-stage disease and honeycombing despite treatment. According to some reports, 5-year mortality can range between 33 and 50% ref.
Transformation to lymphoma can occur, particularly in a patient with monoclonal gammopathy or hypogammaglobulinemia 8. Corticosteroids have been successfully trialled 1.
Complications
Approximately 5% of cases may transform into lymphoma 15.
History and etymology
It was originally classified as an idiopathic interstitial pneumonia in 1969 by Liebow and Carrington.






 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.