
Spinocerebellar ataxias (SCAs) comprise a large and expanding group of neurogenetic diseases characterized by degeneration of the spinal cord and cerebellum. There are over 50 individual spinocerebellar ataxias, referred to sequentially as SCA1, SCA2, ..., etc., roughly in their order of discovery and genetic characterization 2.
On this page:
Epidemiology
As a group, spinocerebellar ataxias are rare. The most common forms vary depending on geographic region, but generally include SCA1, SCA2, SCA3, and SCA6 3,4. Most present in early adulthood, but there is considerable variability 4.
Clinical presentation
The clinical presentation varies depending on the exact type of spinocerebellar ataxia. Common clinical features among these conditions, unsurprisingly, include progressive cerebellar ataxia 4. However, variably patients may have movement disorders, spasticity, sensory symptoms, and ocular anomalies 4.
Pathology
Spinocerebellar ataxias are inherited conditions 4. Many, including the common types, are repeat expansion disorders 4. Among the repeat expansion spinocerebellar ataxias, the common forms (i.e. SCA1, SCA2, SCA3, and SCA6) are specifically CAG repeat expansion disorders 4.
Radiographic features
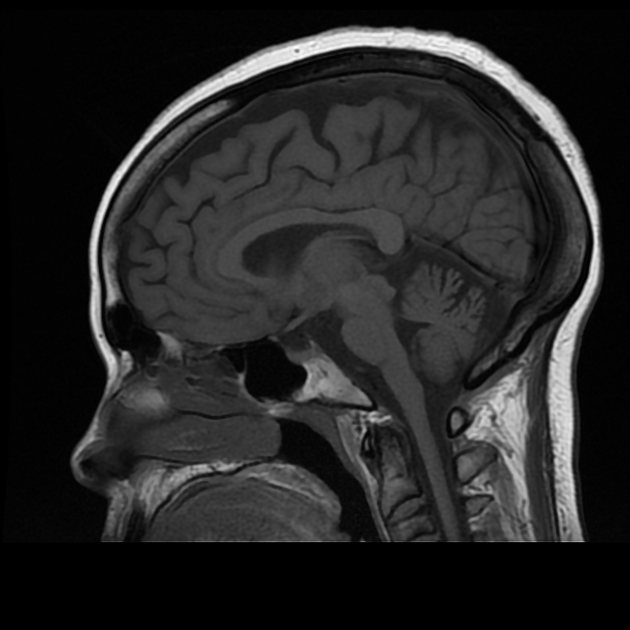
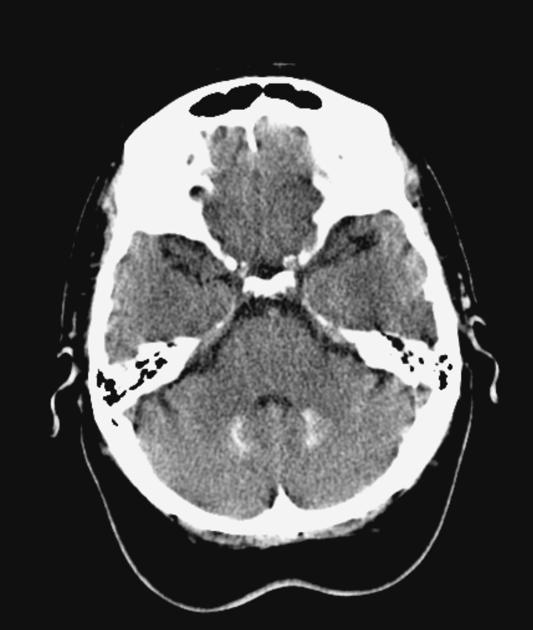
Although individual spinocerebellar ataxias vary in morphological changes, atrophy of the cerebellum is a relatively constant finding. Extracerebellar regions are also affected, depending on the specific SCA 1.
Treatment and prognosis
Management is generally supportive, with no disease-modifying treatments available 4. Among therapies trialed, riluzole may have modest benefit 5, but this benefit has not been replicated across all forms of spinocerebellar ataxia 6.


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.