
Transthyretin (ATTR) amyloidosis is a form of systemic amyloidosis characterised by the misfolding, aggregation and deposition of transthyretin-related (TTR) protein in various organs 1-6. This can occur in the following two forms namely in the setting of a genetically normal transthyretin-related protein also referred to as wild-type transthyretin (ATTRwt) amyloidosis, and in the context of an amyloidogenic mutation of transthyretin, which is then termed variant transthyretin (ATTRv) amyloidosis 1-5.
The genetically normal form or wild type mainly affects the heart and soft tissues. For the hereditary or variant forms cardiomyopathy-predominant, polyneuropathy-predominant and mixed phenotypes are distinguished 1-5 and additionally a type with occuloleptomeningeal involvement 5,6.
On this page:
Terminology
Wild-type ATTR amyloidosis has been formerly known as “senile” or “age-related” ATTR amyloidosis 1-7.
Variant ATTR amyloidosis has been also known as “hereditary”, “familial” or “mutant” ATTR amyloidosis 5,6 with polyneuropathy-predominant variants being also known as familial amyloid polyneuropathy (FAP) 6.
Epidemiology
The true incidence and prevalence of ATTR amyloidosis and transthyretin amyloid cardiomyopathy are currently unknown due to population-based and regional variations of the genetic variant, and also due to low awareness and fragmented knowledge of the disease, resulting in underdiagnosis due to phenotypic heterogeneity and overlap with other clinical conditions 1-9.
Wild-type transthyretin amyloidosis has been increasingly recognised as a cause of heart failure in the elderly 1-9. Misfolding and aggregation seem to increase with age 10-12 with autopsy series indicating TTR amyloid deposits in up to 25% of patients over the age of 80 years 10-12 (however, not all of them to a clinically manifesting degree). The median age at the time of diagnosis is approximately 75 years and men are far more commonly affected 1-4.
Variant transthyretin-related is the most common form of hereditary amyloidosis with variable phenotypes and varying ages at the onset of the disease 1-4. The most common mutation is p.Val50Met (formerly Val30Met) 3,4 with an early onset phenotype mainly seen in Portuguese, Brazilian, Japanese and Swedish populations and a late onset phenotype found worldwide. Other common variants are seen in North America and Western Africa in African-American populations in the Irish and other European regions such as Denmark and Italy 4,5.
Associations
Transthyretin amyloidosis might be associated with the following clinical conditions 1-6:
heart failure with preserved ejection fraction (6-30%) 4,14
aortic stenosis – low-flow-low-gradient (up to 16%) 15
peripheral neuropathy 16
-
wild-type ATTR 17-19:
-
variant ATTR 16,20:
polyneuropathy
autonomic dysfunction
gastrointestinal problems
vitreous opacities
leptomeningeal involvement
Diagnosis
The diagnosis can be established histopathologically and consecutive amyloid typing (e.g. by determination of precursor protein by mass spectrometry). This can be done by tissue biopsy of the affected organ (e.g. the heart or peripheral nerves) or at other sites including the gastrointestinal mucosa, abdominal fat pad and salivary glands 3,12,13,16.
Alternatively, the diagnosis can be established noninvasively in the following context 12,13,21:
positive bone scintigraphy (99mTc-PYP, 99mTc-DPD, , 99mTc-HMDP) with a Perugini grade 2 or 3
characteristic findings on echocardiography or cardiac magnetic resonance
negative serum-free light chains as well as serum and urine immunofixation tests
In addition, genetic testing is part of the diagnostic work-up 12.
Clinical presentation
Due to its phenotypic heterogeneity symptoms are often variable and unspecific leading to a delay in diagnosis 3-5.
In wild-type transthyretin-related amyloidosis, presenting symptoms are often cardiac in origin, including fatigue, dizziness, dyspnoea and exercise intolerance 1.
In variant transthyretin-related amyloidosis, a cardiac and a neurological phenotype are distinguished, with the neurological phenotype showing symptoms of peripheral neuropathy and autonomic dysfunction, the latter potentially causing orthostatic dysfunction and syncope 4,12,13.
Other extracardiac symptoms include musculoskeletal or gastrointestinal symptoms, erectile dysfunction or unexplained weight loss 1-3,12,13.
In addition to the clinical symptoms the following tests might be abnormal 2-5,12,13:
NT-proBNP or troponin levels might be elevated, increase over time and are thus included in staging systems 2-5
ECG might show decreased limb QRS voltage (≤5 mV)
abnormal nerve conduction studies and autonomic testing
Complications
Complications include the following 3-5,13:
conduction anomalies
Pathology
Transthyretin-related amyloidosis is characterised by misfolding, aggregation and deposition of transthyretin-related protein as insoluble amyloid fibrils in the extracellular space of various organs including the heart, vasculature, the peripheral and autonomic nervous system, the soft tissues and gastrointestinal tract 1-4. The amyloid fibrils are formed by the transport protein transthyretin (TTR) after dissociation of its tetrameric form into monomers and subsequent misfolding, either as a result of favourable conditions like oxidative stress or ageing or in the context of an amyloidogenic mutation facilitating the process 3,5 Transthyretin is mainly synthesised in the liver and to a lesser extent in the choroid plexus and by retinal pigment epithelial cells 3,5.
Subtypes
Transthyretin-related amyloidosis features the following subtypes 1:
wild-type (ATTRwt): genetically normal protein with an intrinsic tendency to form amyloid under certain conditions
variant from (ATTRv): amyloidogenic mutation promoting the formation of amyloid fibrils
Microscopic appearance
Amyloidosis is confirmed histopathologically by Congo red positivity of the extracellular amyloid deposits showing their fibrillar ultrastructure associated with apple green birefringence under polarised light 1-5.
Immunophenotype
Transthyretin-related protein can be detected immunohistochemically in affected tissues using an anti-transthyretin antibody 18. Immunohistochemistry and related methods such as immunofluorescence and immunoelectron microscopy are considered alternative methods to mass spectrometry, they are however less sensitive and less specific 1.
Genetics
Genetic sequencing and counselling are considered an essential part of the evaluation of ATTR amyloidosis for the following reasons 1,12:
differentiating wild-type ATTR from variant ATTR
identification of relatives at risk
potentially influence of treatment strategies and prognosis
The tetrameric serum protein transthyretin is encoded by the TTR gene found on chromosome 18q12.1 and spanning four exons 4. More than 140 different mutations are known 4 with the more common forms causing variant ATTR being p.Val142Ile (formerly Val122) common in African-Americans, p.Val50Met (formerly Val30) common among Portuguese, Japanese and Swedish populations) and p.Thr80Ala (formerly Thr60Ala) common among the Irish people 4,12.
Radiographic features
Imaging features of cardiac amyloidosis in general include the following 12,13,22-24:
-
unexplained increased wall thickness of the following structures:
left ventricular myocardium: often asymmetric
right ventricular free wall
interatrial septum and right atrial wall
biatrial enlargement
Additionally 22-24:
See also: cardiac amyloidosis
Ultrasound
Echocardiography
Echocardiography serves as a first-line imaging tool in the evaluation of cardiac involvement of amyloidosis. Echocardiographic criteria for cardiac amyloidosis in general include an increased wall thickness (≥12 mm) and other characteristic features such as 12,13,22:
decreased tissue Doppler s’, e’ and a’ wave velocities (<5 cm/s)
decreased global longitudinal left ventricular strain with apical sparing 22
Other common features include a ‘speckled pattern’ or ‘granular sparkling’ of the myocardium as well as biatrial enlargement 2,13.
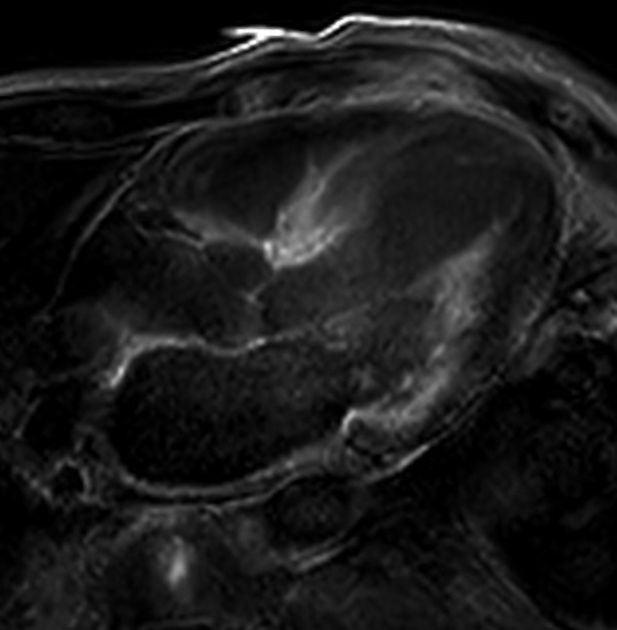
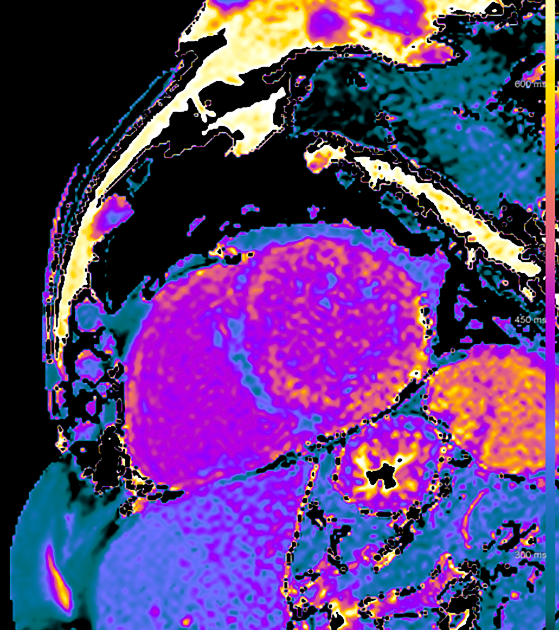
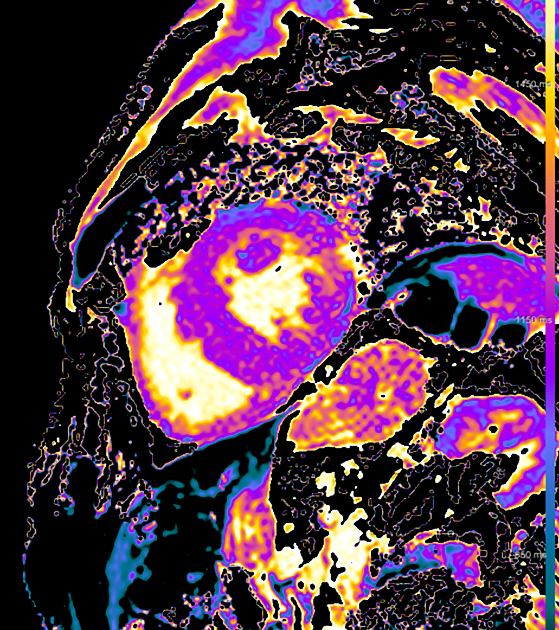
MRI
Cardiac MRI usually shows a significantly increased left ventricular mass, diffusely increased native T1 and very high ECV values on myocardial mapping and a variably diffuse to transmural non-ischaemic late enhancement pattern as well as abnormal blood pool gadolinium kinetics 12,13.
Signal characteristics
IRGE/PSIR: often diffuse late enhancement with right ventricular involvement 25
T1 mapping: increased
ECV: markedly increased (often ≥50%) 12,13
It is worth mentioning that a reliable differentiation between ATTR and AL on cardiac MRI is not possible 12,13,22,23, still, MRI features favouring ATTR amyloidosis over AL include 24,25:
asymmetrical wall thickening
high native T1 mapping values but lower than in AL amyloidosis
very high extracellular volume values >50%
high left and right ventricular wall thickness
high left ventricular mass >100 g/m2
late gadolinium enhancement is often more diffuse with right ventricular involvement and a QALE score >13
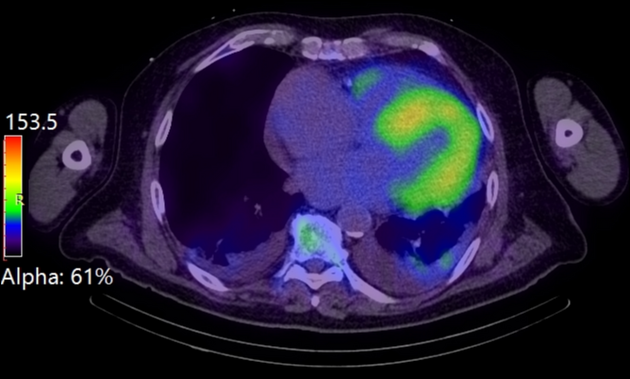
Nuclear medicine
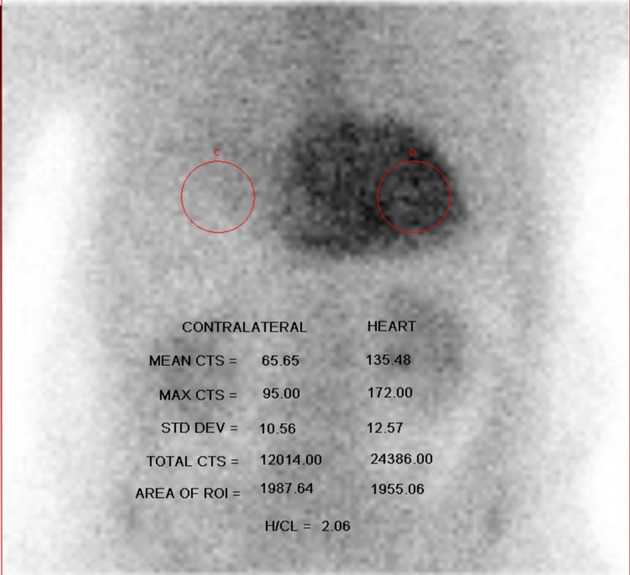
Bone scintigraphy with Perugini grading is usually positive in the setting of cardiac ATTR-amyloidosis with a Perugini grade of 2 or 3, pointing to ATTR amyloidosis 12,13,22.
However, in a significant percentage (>10%) of patients with AL amyloidosis bone scintigraphy has been also reported to be positive as well as in different other forms of amyloidosis 7,8. Also, bone scintigraphy can be falsely positive in recent myocardial infarction (< 4 weeks), hydroxychloroquine cardiac toxicity as well as valvular or annular calcifications 13.
In addition, it can be negative in some ATTRv mutations 13.
Radiology report
The radiology report should include a description of the following:
morphology and functional analysis
left ventricular wall thickness
abnormal strain patterns (if measured)
valvular thickening and atrial enlargement (if present)
MRI
-
myocardial tissue properties including:
presence, pattern and distribution of late gadolinium enhancement
alteration of blood-pool gadolinium kinetics
abnormal T1 and T2 mapping and ECV values (if performed)
Treatment and prognosis
Management includes guideline-directed therapy of cardiovascular symptoms, prevention and treatment of complications, new targeted therapeutic options aimed at preventing the dissociation of TTR molecules into amyloidogenic fragments by stabilising their circulating forms (stabilising molecules) and interfering with their production (genetic silencers) as well as supportive measures 5,12,13.
The targeted therapy regimen depends mainly on the ATTR subtype (hereditary or wild type) and thus on the organ involvement of the disease, i.e. the presence of cardiomyopathy or polyneuropathy or both 7 and currently includes the following agents 12,13:
-
TTR tetramer stabilisers: tafamidis, duflunisal, acoramidis
in the setting of cardiomyopathy
-
gene silencers: partisiran, inotersen
in the setting of polyneuropathy
Liver transplantation was the main treatment option in variant ATTR before the introduction of stabilising and genetic silencing agents 1,6.
The prognosis of transthyretin-related amyloidosis varies with the subtype, mutation and phenotype, the time of diagnosis as well as the presence of complications ranging from less than 3 years in patients with cardiomyopathy-predominant phenotypes up to 8 or 10 years in polyneuropathy-predominant variants and approximately 5 years in the setting of wild-type cardiac amyloidosis 2,5.
The prognosis of transthyretin amyloidosis is generally better than in light chain amyloidosis, especially when the heart is involved 6,12. The extent of late gadolinium enhancement and extracellular volume are considered predictors in cardiac amyloidosis 23,26.
History and etymology
The term amyloid was first used for medical purposes by Rudolf Virchow in 1854 initially considering amyloid starch-based extracellular deposits 27-29.
Senile amyloidosis was first reported by the Czech physician and pathologist Isidora Soyka in 1876 7,30. Hereditary ATTR amyloidosis was first reported in Portugal by the Portuguese neurologist Corino Andrade in 1952 and later in Japan and Sweden 28,31-33.
Differential diagnosis
The main differential diagnosis of ATTR amyloidosis includes the following 2,12:
other infiltrative cardiomyopathies such as Fabry disease
diabetic polyneuropathy
peripheral polyneuropathies
Practical points
ATTRwt has been considered a cardiac disease, whereas ATTRv has neuro-predominant and cardiac-predominant variants 3
on cardiac MRI, PSIR is an option to better deal with the difficulties of nulling myocardium 26
the abdominal fat pad offers a rather low sensitivity as a biopsy site for ATTR amyloidosis (~45% in ATTRv and ~15% ATTRwt)
alternative biopsy sites for suspected ATTR are the labial salivary gland or the sural nerve, the latter, especially in the setting of polyneuropathy 25
variant (hereditary) forms should undergo regular neurological as well as ophthalmological evaluations 13
follow-up should include yearly echocardiography or cardiac MRI 13


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.