
Autoimmune glial fibrillary acid protein (GFAP) astrocytopathy, or simply GFAP astrocytopathy, is a rare inflammatory central nervous system (CNS) disorder.
On this page:
Epidemiology
Given the rarity of the condition, epidemiological data pertaining to autoimmune GFAP astrocytopathy are not well established. Based on existing case series-level data, it tends to manifest in middle-aged adults (but can also affect paediatric patients 12) and there are conflicting reports regarding any sex preponderance 1,13.
Associations
There may be an association with an underlying tumour or malignancy (e.g. ovarian teratoma (most common), a variety of adenocarcinomas), in approximately one-third of cases 1-6,8.
Clinical presentation
Autoimmune GFAP astrocytopathy has a broad clinical neuropsychiatric and temporal spectrum 1-6,8. The most common phenotypical syndromes are those of meningoencephalitis or meningoencephalomyelitis 1-6,8, which can manifest over an acute, subacute, or less commonly, a chronic time-frame 4,8.
Within those syndromes, a wide range of clinical features may be present, the most common of which include 1-6,8:
headache
neck stiffness
motor and sensory changes (e.g. due to myelopathy)
visual changes (due to optic papillitis and/or optic neuritis)
cerebellar ataxia
autonomic dysfunction
movement disorders (e.g. tremor, chorea, myoclonus)
drug-refractory seizures and epilepsy
bulbar symptoms
psychiatric symptoms (e.g. psychosis)
Additionally, a coryza or flu-like prodrome is also a common clinical feature, being present in 30-67% of patients 4-6.
Pathology
Aetiology
The pathophysiology of autoimmune GFAP astrocytopathy has not been fully elucidated and is not well understood. It is known that GFAP is an intracellular protein that forms an integral component of astrocyte cytoskeletons and also plays a pivotal role in multiple astrocytic functions 4. In autoimmune GFAP astrocytopathy, there are IgG autoantibodies that target GFAP in astrocytes, which leads to an eventual loss of astrocytes, and presumably the resultant clinical phenotype 1-6. These autoantibodies may have a paraneoplastic or parainfectious aetiological basis 1.
Markers
The key marker is the GFAP antibody, which has a higher positive predictive value when present in the CSF than in the serum 5. Additionally, CSF will often have an inflammatory phenotype, with high protein and a pleocytosis 1-6. Co-existing autoantibodies (e.g. NMDA receptor antibody, aquaporin-4 antibody) may also be present, especially in patients with ovarian teratoma 1-6,11.
Microscopic appearance
Typical neuropathological features include 1,3,11:
extensive perimicrovascular inflammation featuring both B and T cells, sparing the vessel itself
microglial activation
Radiographic features
MRI
Radiographic abnormalities of the neuraxis are very common in autoimmune GFAP astrocytopathy 1-6. Imaging appearances usually improve following treatment 13,14.
Brain
Typical findings include:
-
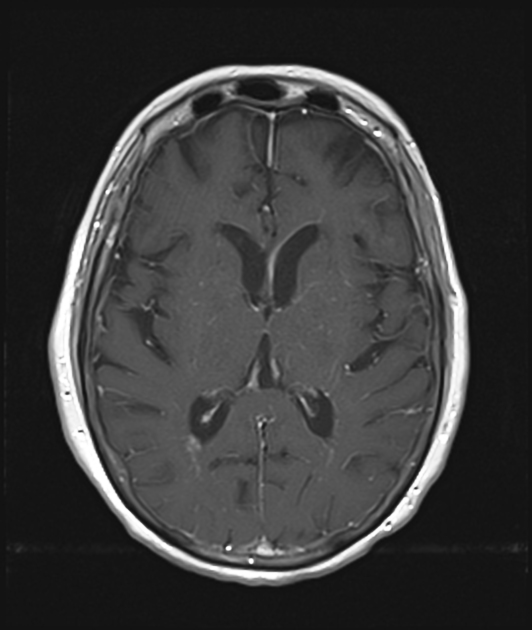
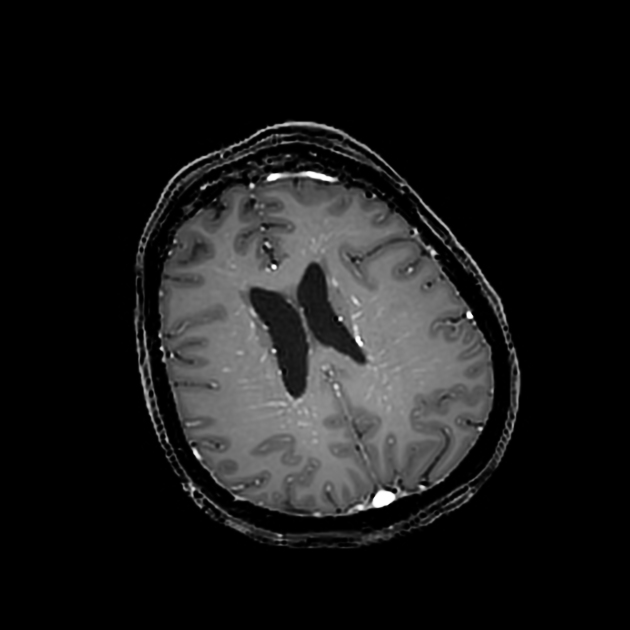
T2/FLAIR: hyperintense, diffuse, confluent, periventricular white matter lesions (present in up to 75% of cases) 1-6,11, sometimes extending into or otherwise involving the cortex, centrum semiovale, thalami, basal ganglia, and/or brainstem 1,3,11,13
in paediatric patients, thalami and basal ganglia may be more likely to be affected 12
DWI: normal 6
-
T1 C+ (Gd): characteristic (present in ~50% of cases) linear, perivascular pattern of contrast-enhancement extending radially from the periventricular surface and through the cerebral white matter, although can also be seen extending through the thalami and basal ganglia 1-6,11,12
other less characteristic patterns of enhancement include leptomeningeal, punctate or periependymal 1,2,4-6,11,13
MR angiogram: normal 6
Notably, in patients with a predominantly cerebellar ataxia phenotype, diffuse cerebellar atrophy may be the only radiographic feature 4. Furthermore, despite visual changes being a relatively common feature of autoimmune GFAP astrocytopathy, optic nerves often appear normal on imaging 9, but rarely there can be features of optic neuritis and optic perineuritis 13.
Spinal cord
Typical findings, most commonly in the cervicothoracic spinal cord, include:
-
T2: hyperintense longitudinally extensive spinal cord lesion(s), often pericentrally located 1-6,13,14
lesions are described as having “hazy” or indistinct borders 6,13
may also have focal cord atrophy 3
T1 C+ (Gd): may have contrast-enhancement, which is usually leptomeningeal or of the central canal 1-6,13,14
Nuclear medicine
PET-CT
FDG PET-CT of the neuraxis may reveal increased FDG uptake indicative of hypermetabolism, often corresponding with regions of signal abnormality on MRI brain and/or spinal cord 1-3,5,6.
Treatment and prognosis
Autoimmune GFAP astrocytopathy is typically steroid-responsive 1-6. In cases that are steroid-refractory, often in the setting of concurrent underlying malignancy or another co-existing autoantibody, there may be response to other immunosuppressive agents (e.g. rituximab, mycophenolate mofetil, azathioprine, cyclophosphamide, intravenous immunoglobulin) 5.
The disease is most often monophasic, however, may have a relapsing course in approximately 20% of patients 6. Overall, the prognosis is generally good 6.
History and etymology
Autoimmune GFAP astrocytopathy was first described by Boyan Fang, Vanda Lennon, and colleagues from the Mayo Clinic in their 2016 seminal paper 2. It is thought that some cases previously labelled as 'nonvasculitic autoimmune inflammatory meningoencephalitis' may actually have been autoimmune GFAP astrocytopathy 3.
Differential diagnosis
neurosarcoidosis 7,11
anti-vimentin encephalomyelitis 15


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.