
Primary hyperoxaluria, also referred to as primary oxalosis, is a congenital autosomal recessive disease related to a liver enzyme deficiency leading to massive cortical nephrocalcinosis and renal failure.
Please refer to secondary oxalosis for a discussion on the acquired form of hyperoxaluria.
On this page:
Epidemiology
Hyperoxaluria is an autosomal recessive disorder and is more common in Mediterranean countries. The most common subtype is primary hyperoxaluria type 1, which is responsible for approximately 80% of cases 3.
Clinical presentation
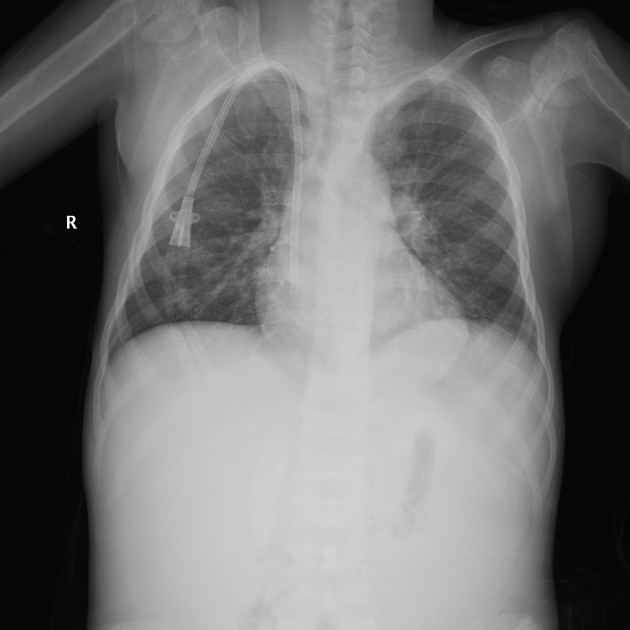
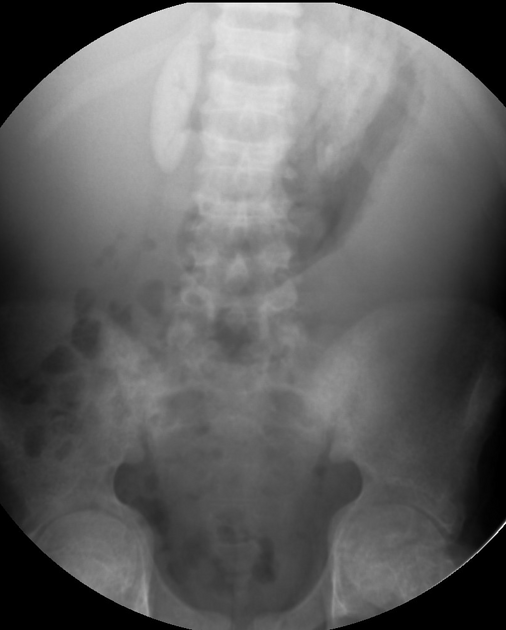
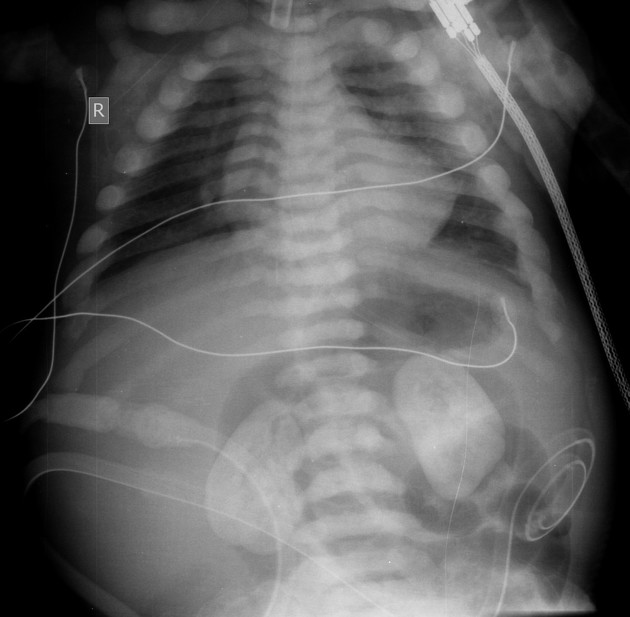
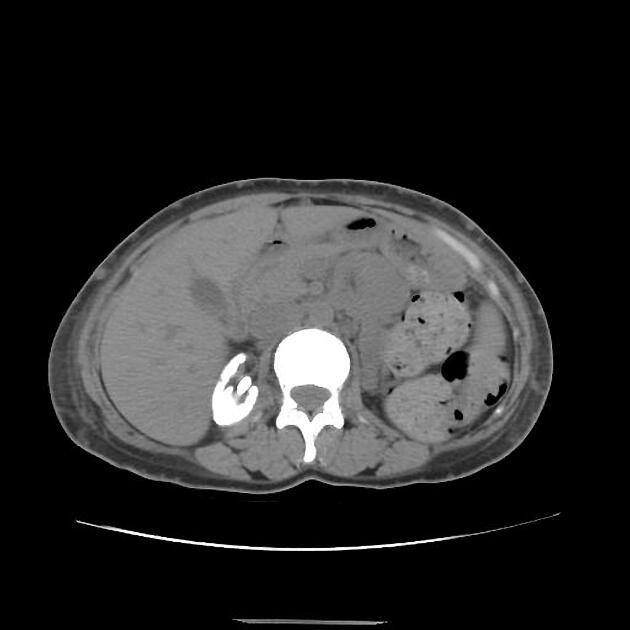
The typical presentation is nephrolithiasis and global (cortical and medullary) nephrocalcinosis at an early age.
Pathology
It has three types due to the defects in the gene that encodes the following enzymes:
glyoxylate aminotransferase
glyoxylate reductase/hydroxypyruvate reductase
liver-specific mitochondrial 4-hydroxy-2-oxoglutarate aldolase enzyme
Radiographic features
Plain radiograph
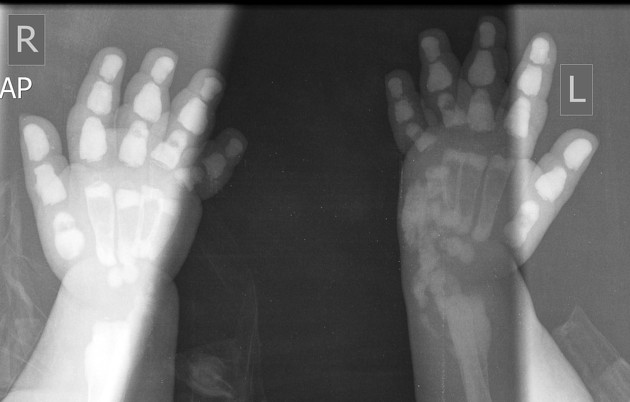
Plain radiographs demonstrate osteosclerosis as well as osteopenia. Other skeletal abnormalities include fine transverse lines of increased bone density located in areas of rapid growth, submarginal metaphyseal lucency, wide translucent metaphyseal zone with sclerosis of the adjacent diaphysis, and metaphyseal “waisting” or focal narrowing.
Diagnosis by usually by bone (iliac crest) biopsy.
Treatment and prognosis
Treatment is with a combined liver and renal transplantation (the liver transplant is to rectify the enzyme deficiency, not because of liver failure).
The pyridoxine supplementation may be used in conjunction with combined liver-kidney transplantation to enhance the alternative pathway of glyoxylate metabolism to glycine and, therefore, reduce the amount of glyoxylate available for conversion to oxalate.
An RNA interference (RNAi) treatment has begun trials that targets the mRNA of glycolate oxidase, an enzyme in the pathway leading to oxalate 4. As in primary hyperoxaluria type 1, enzyme deficiency leads to high oxalate levels. The siRNA lumasiran reduces oxalate levels to normal or nearly normal levels within weeks, and results are consistent for long-term treatment. Lumasiran is given in monthly loading doses and the dosage is weight-dependent which allows treatment from birth.


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.