
X-linked adrenoleukodystrophy is an inherited metabolic peroxisomal disorder and one of the more common leukodystrophies in both children and adults. It is characterised by lack of oxidation of very long chain fatty acids (VLCFAs) that results in severe inflammatory demyelination typically of the periventricular deep white matter with posterior-predominant pattern and early involvement of the splenium of the corpus callosum and parietal white matter changes. Most patients will demonstrate characteristic MRI brain findings.
On this page:
Epidemiology
The estimated incidence of adrenoleukodystrophy is 1:20,000-50,000. Due to its X-linked inheritance, it classically affects young males, although carrier females can be affected. Although most cases are diagnosed in childhood, a significant proportion of cases manifest in young adults (typically late 20s) 3,11,12 and thus adrenoleukodystrophy is one of the most common adult-onset leukodystrophies 15.
Clinical presentation
The presentation will depend on the phenotype which in turn depends upon the age (see below). Some individuals can be asymptomatic.
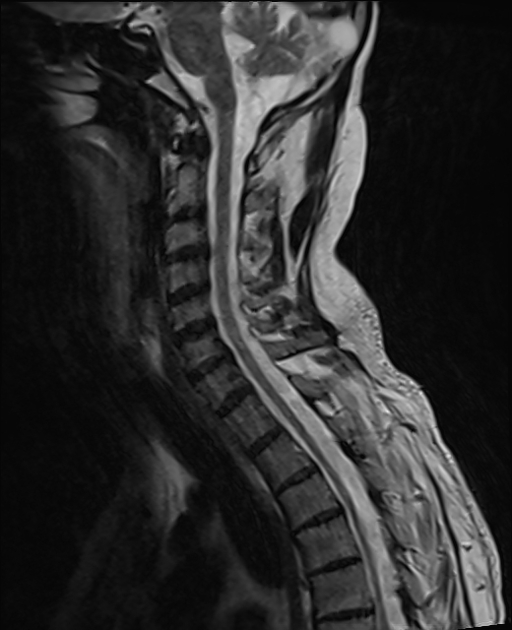
As a general rule, most children will have prominent supratentorial involvement, whereas adults will have more pronounced spinal cord involvement (adrenomyeloneuropathy) 15.
Phenotypes
Up to eight phenotypes have been described but the three main types in males are 3,8,11,12,15:
-
cerebral adrenoleukodystrophy (40%)
presents after 2.5 years of age (typically at 4-8 years)
progressive impairment motor and cognitive function, vision and hearing
when adults present with cerebral involvement presentation is often neuropsychiatric prior to other manifestations (ataxia, seizures, dementia) 15
-
adrenomyeloneuropathy (35-40%)
most common adult presentation
progressive paraparesis, sphincter dysfunction, sexual dysfunction, and adrenocortical deficiency
spinal cord involvement with few if any brain abnormalities early in the disease
-
Addison disease only (~10%), i.e. primary adrenocortical deficiency
no symptomatic leukodystrophy
Approximately 20% of female carriers will be affected, although onset is late (>35 years) with milder paraparesis 11.
Pathology
The conditions result from the accumulation of very long-chain fatty acids (VLCFAs) due to genetic deficiency in the peroxisomal oxidation of fatty acids. This is thought to result from a mutation in the ABCD1 gene located on chromosome Xq28 that encodes the protein adenosine triphosphate-binding cassette transporter embedded within the peroxisomal membrane 5,11,15.
The affected cerebral white matter is typically split into three different zones (also referred to as Schaumberg zones 3, 2, and 1, respectively):
central (inner) zone: irreversible gliosis and scarring
intermediate zone: active inflammation and breakdown of the blood-brain barrier
peripheral (outer) zone: leading edge of active demyelination
Markers
markedly elevated VLCFA concentrations in plasma and cultured skin fibroblasts 11,13
Radiographic features
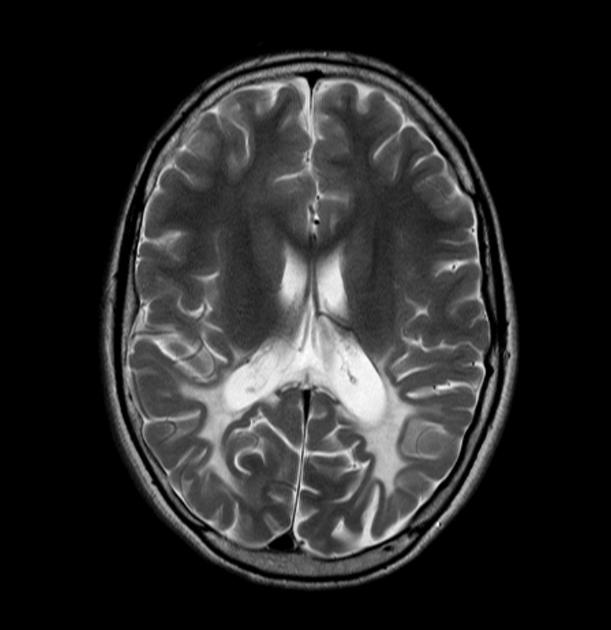
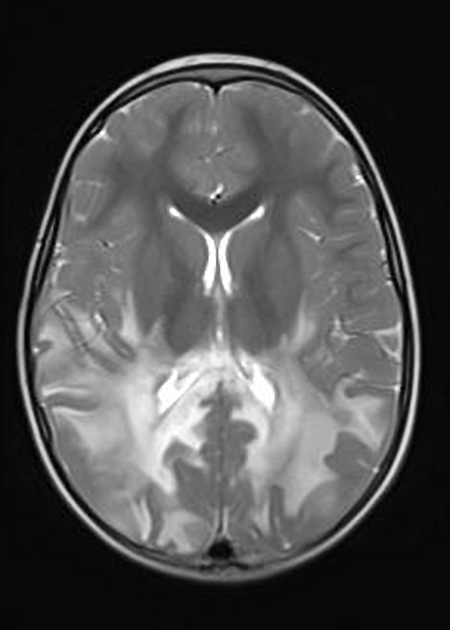
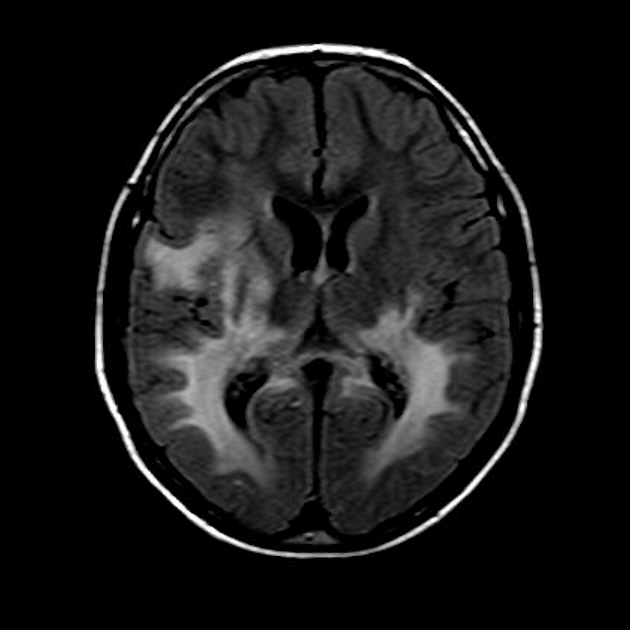
MRI
Abnormal MRI findings precede clinical findings in all forms of adrenoleukodystrophy 13. Distribution of involvement results in different clinical forms of the disease.
Location
Loes et al. 10 described five different MRI brain patterns of adrenoleukodystrophy based on the involved anatomic locations and MRI patterns of progression:
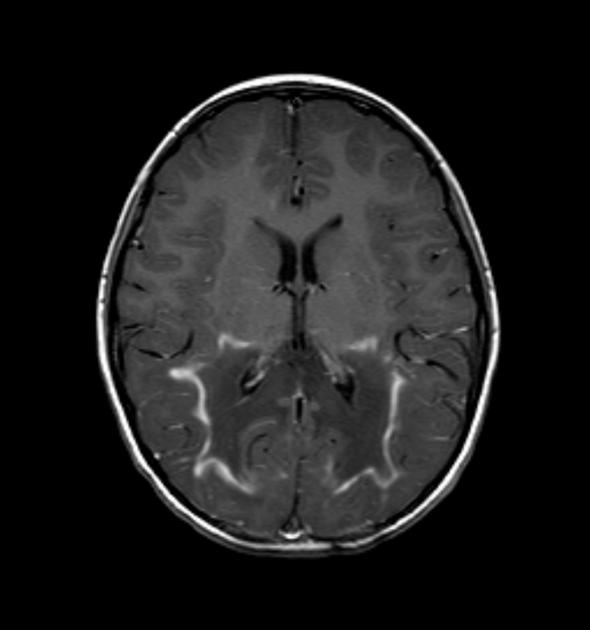
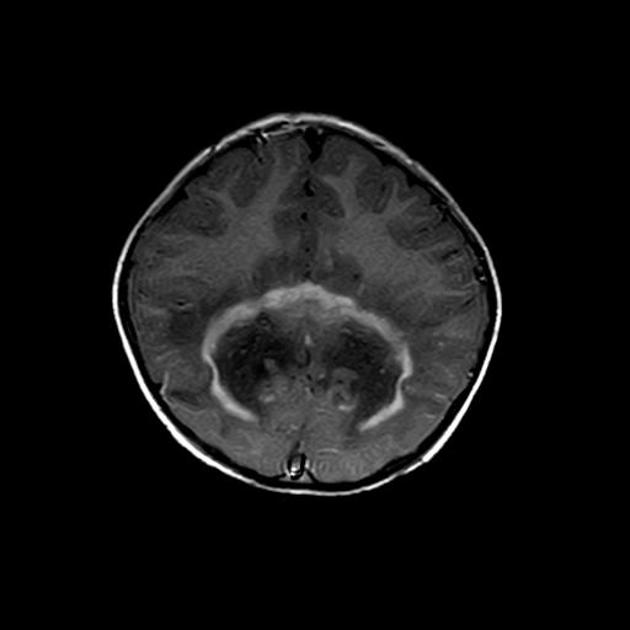
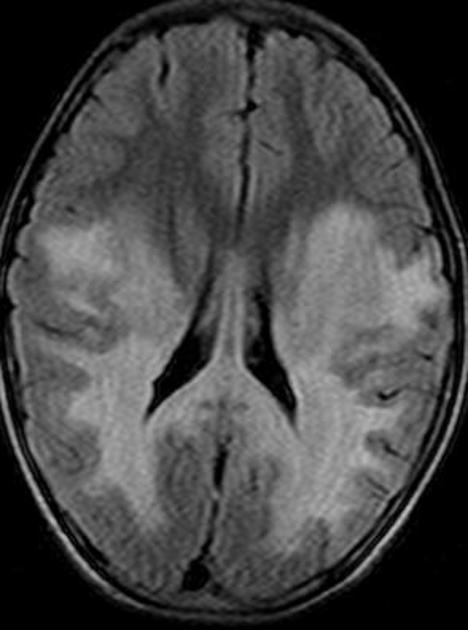
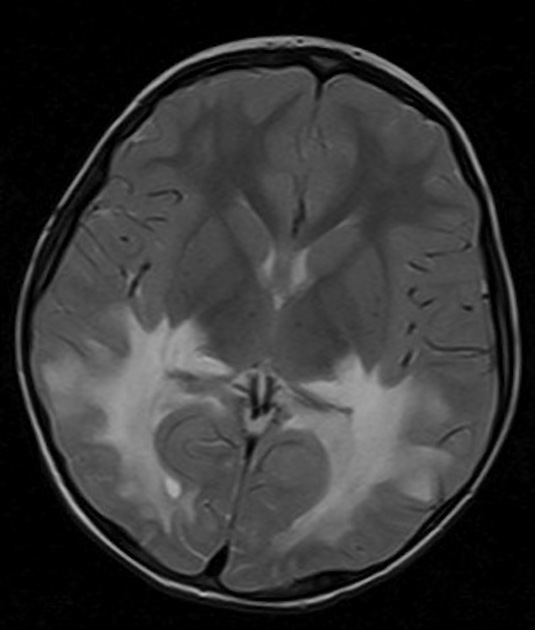
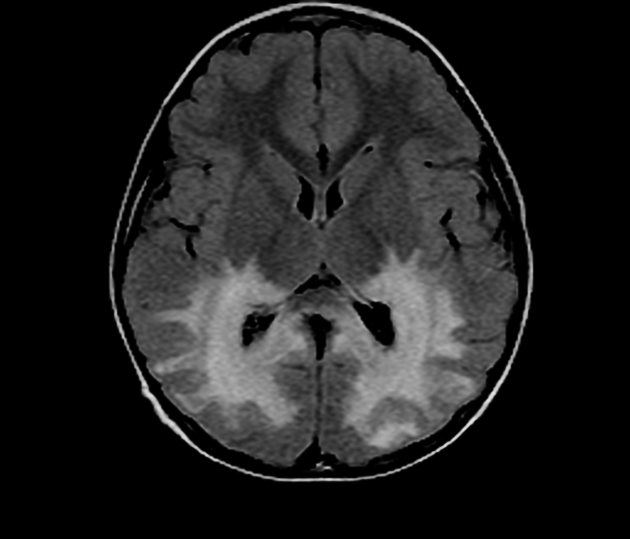
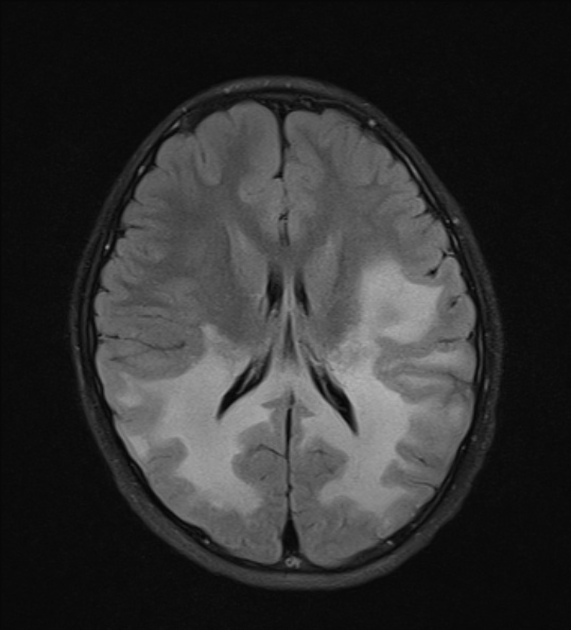
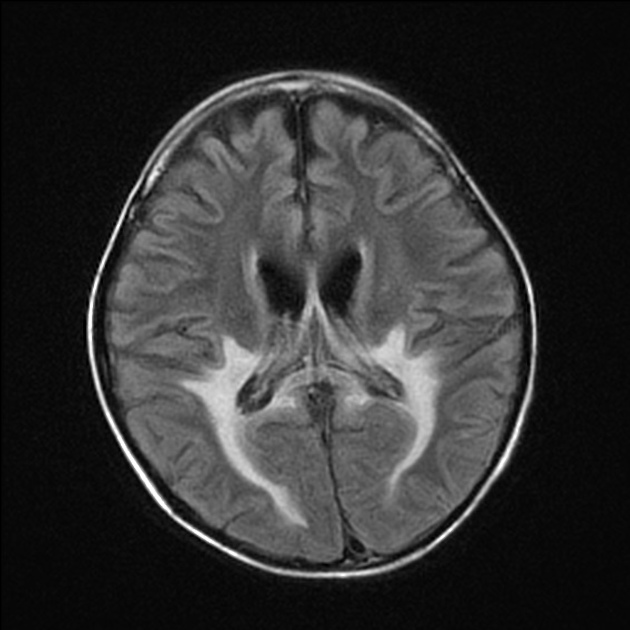
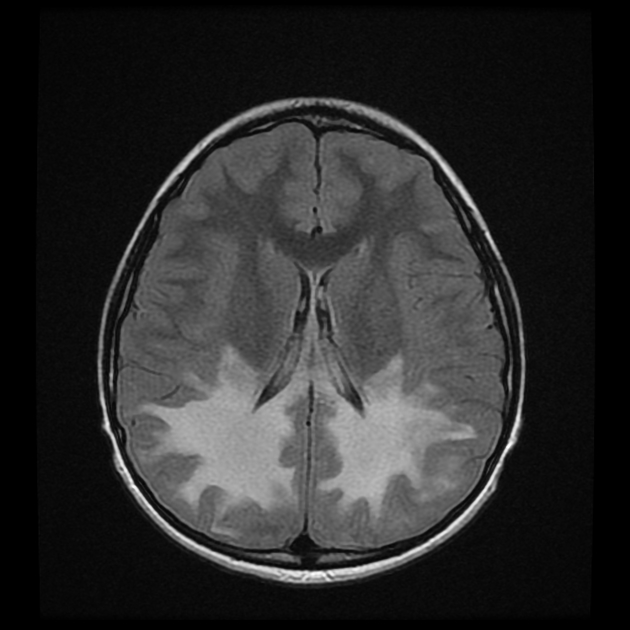
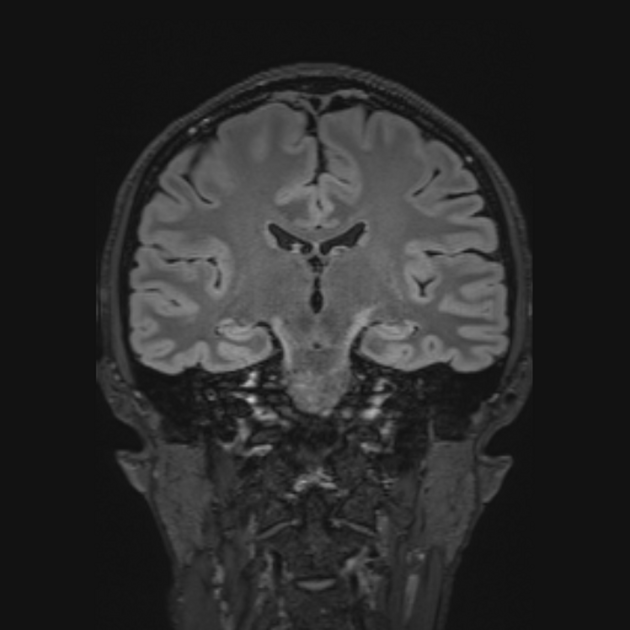
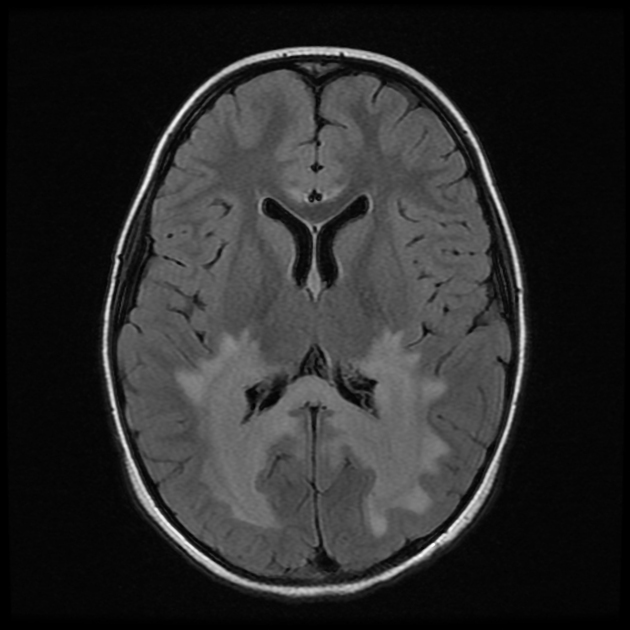
deep white matter in the parieto-occipital lobes and splenium of the corpus callosum (66% of cases, chiefly in children); may include lesions of the visual and auditory pathways
frontal lobe or genu of the corpus callosum (15.5%, mostly in adolescents)
frontopontine or corticospinal projection fibres (12%, mostly in adults)
cerebellar white matter (1%, mostly in adolescents)
combined parieto-occipital and frontal white matter (2.5%, mostly children)
There tends to be cortical and subcortical U-fibre sparing.
Spinal cord involvement is pronounced in the adrenomyeloneuropathy form of the disease, particularly affecting the thoracic segment 15.
Signal intensity
Signal changes can vary according to the zonal distribution within the affected white matter.
-
T1
central zone: hypointense
intermediate zone: hyperintense
peripheral zone: hypointense
-
T1 C+ (Gd)
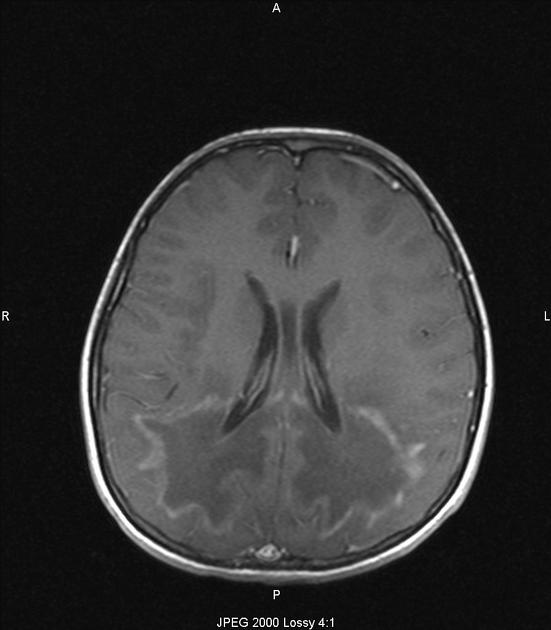
enhancement is seen in around 50% of cases according to one study and is thought to be associated with disease progression 6
with contrast infusion, serpiginous, garland-shaped enhancement may be visible in the anterior most periphery of the lesions 7
-
T2
central zone: markedly hyperintense
intermediate zone: isointense to hypointense
peripheral zone: moderately hyperintense
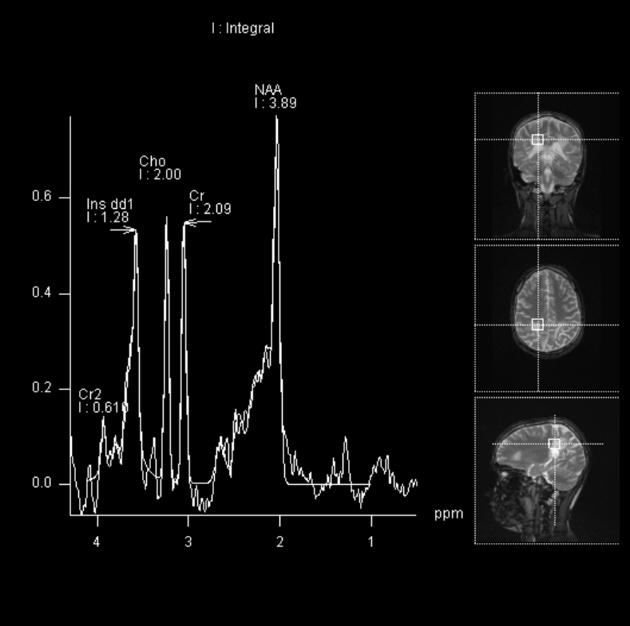
MR spectroscopy
Spectroscopy may show evidence of neuronal loss manifested by a decrease in the NAA peak and an elevation in the lactate peak 2,14.
Treatment and prognosis
Haematopoietic stem cell transplantation is thought to be a favourable therapeutic option in the early stages of the disease. In adults, leriglitazone may have benefit in slowing progression of early cerebral disease 16. Steroid replacement should also be given in patients with adrenocortical insufficiency.
The pattern of involvement also determines prognosis, with combined frontal and parieto-occipital usually heralding rapid disease progression, whereas isolated cerebellar or corticospinal tract involvement generally having slower progression 15.
The degree of contrast enhancement at the edge or white matter involvement is also predictive of disease progression 15.
History and etymology
It is thought to have originally been described by Siemerling and Creutzfeldt in 1923 1.
Differential diagnosis
Differential considerations broadly include many other leukodystrophies. In infants/neonates peroxisomal acetyl CoA oxidase deficiency shares many similarities.


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.