
Aicardi syndrome is a rare severe developmental disorder. It results from an X-linked genetic defect that is fatal in males and, therefore, only manifests in females (except for rare 47, XXY cases).
On this page:
Terminology
Aicardi syndrome is distinct from Aicardi-Goutieres syndrome, although both are named after Jean Aicardi (see below).
Epidemiology
Due to its rarity, epidemiological data is limited. However, its incidence has been postulated at ~1/100000, with a prevalence of over 850 cases in the US and 4,000 globally. No ethnicity bias has been found 5.
Diagnosis
Aicardi syndrome is diagnosed clinically, supported by neuroimaging and EEG findings and sometimes genetic studies. The classic triad must be present 6:
agenesis of the corpus callosum
chorioretinal lacunae
infantile spasms or early-onset epilepsy
Neuroimaging features (see below) and EEG findings are supportive. In some cases speficic mutation may be identified, however, there is no single confirmatory test, and most cases arise from de novo mutations.
Clinical presentation
The typical presentation in infancy is with a triad of:
infantile spasms: salaam seizures with typical bowing of the head
corpus callosal dysgenesis: most consistent feature
distinctive chorioretinal lacunae: essentially pathognomonic
Pathology
Although in some instances mutations in specific genes on the X chromosome have been identified (e.g. TEAD1 and OCEL1) these are heterogeneous 7.
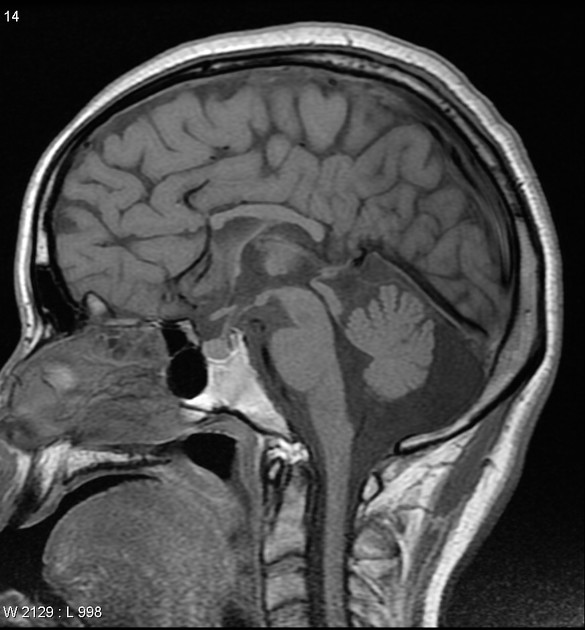
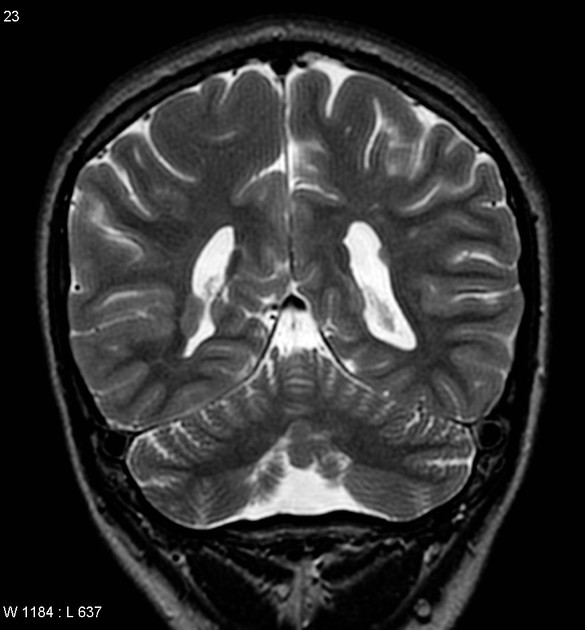
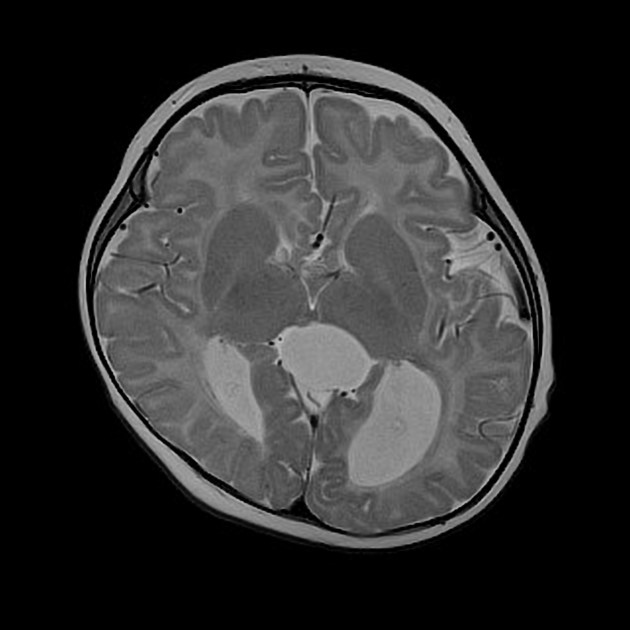
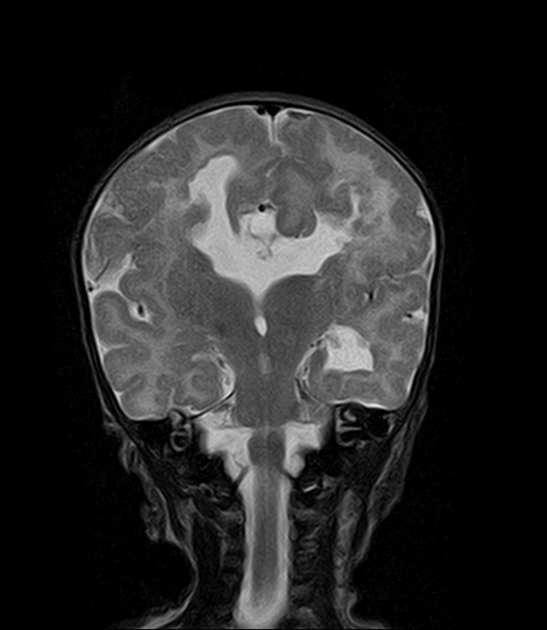
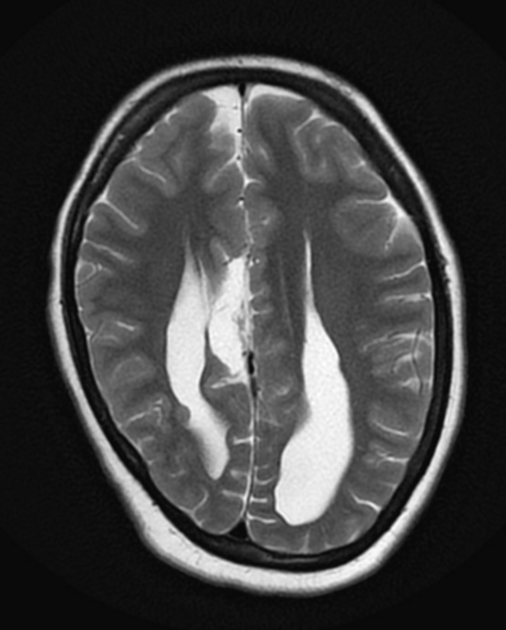
Radiographic features
Characteristic malformations affect the brain, spine and eyes and include:
-
brain
asymmetry of cerebral hemispheres
-
posterior fossa abnormalities (95%), including Dandy-Walker continuum:
posterior fossa cyst
tectal enlargement
polymicrogyria (predominantly frontal and perisylvian)
-
intracranial cysts (diameters range between 1.0-5.0 cm)
midline interhemispheric: 81%
intraventricular: 29%
parenchymal: 10%
extra-axial: 8%
-
ocular
-
spine and ribs
scoliosis: 1/3 cases 5
abnormal costovertebral articulations
History and etymology
First described in 1961 by Jean Francois Marie Aicardi (1926-2015), a French neuropaediatrician 2.


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.