
Charcot-Marie-Tooth (CMT) disease, also known as hereditary motor and sensory neuropathy (HMSN), refers to a heterogeneous group of inherited peripheral neuropathies rather than a single clinical entity 9.
On this page:
Epidemiology
The prevalence of CMT has been reported at ~45 cases (range 10-82) per 100,000 people with significant regional variation 4,10.
Clinical presentation
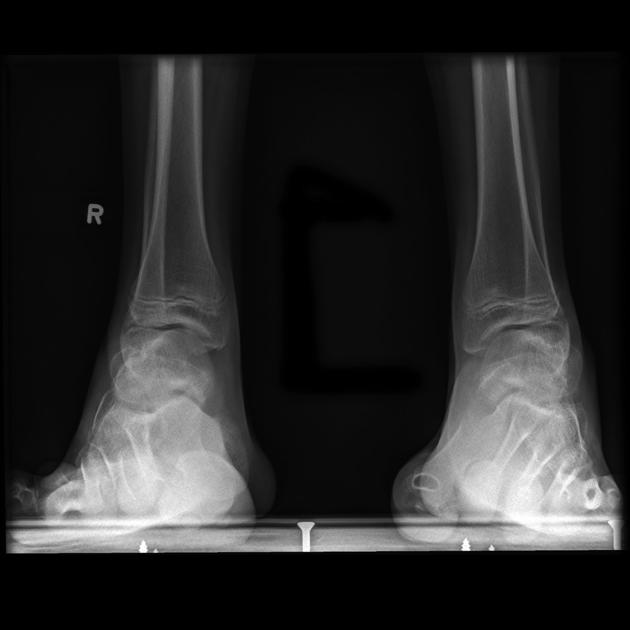
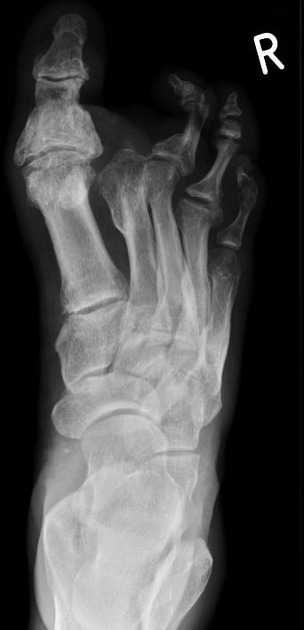
Signs and symptoms usually become evident in childhood. Typically, this starts in the lower limbs with weakness, atrophy, and deformity and later affects the upper limbs. It rarely involves the more proximal musculature or the cranial nerves. Sensory changes are present but usually to a lesser degree 5. Foot deformities include cavovarus (high-arch) and claw-toe deformities ref. Patients can present with scoliosis ref.
Pathology
Charcot-Marie-Tooth disease is not a single condition but a cluster of heterogeneous peripheral nerve disorders with more than 100 causative genes 9.
Most frequently, it is autosomal dominant in inheritance (although this is variable) ref.
Classification
Due to next generation sequencing, the types and subtypes of Charcot-Marie-Tooth disease and their classification are in flux (c. 2023) 9.
Classification is based on nerve-conduction studies and neuronal pathology divided into demyelinating and non-demyelinating forms 5:
Demyelinating forms
Reduced nerve conduction velocity (<38 m/sec) in upper limb nerves and myelin abnormalities on biopsy (e.g. onion bulb formation). This includes:
-
CMT type 1
most common 4,5
autosomal dominant inheritance, most commonly due to over-expression of peripheral myelin protein 22 (PMP22 gene) 5
duplication of chromosome 17
repeated cycles of demyelination and remyelination result in a thick layer of abnormal myelin around the peripheral axons
this form of CMT disease is a disorder of peripheral myelination
these changes cause what is referred to as an onion bulb appearance
-
CMT type 4
autosomal recessive inheritance 5
Axonal forms
Preserved or mildly affected nerve conduction velocity (>38 m/s) and nerve biopsy evidence of degeneration and regeneration 5.
-
CMT type 2
peripheral neuropathy through direct axonal death and Wallerian degeneration
autosomal dominant inheritance 5
most commonly due to a mutation in the mitofusin 2 gene (MFN2) 5
Other forms
pure motor neuropathy (dHMN) with sparing of sensory nerves
pyramidal involvement (CMT type 5)
optic nerve involvement (CMT type 6)
-
CMT type 3 (also known as Déjerine-Sottas disease)
characterized by infantile onset, resulting in severe demyelination with delayed motor skills, it is much more severe than CMT type 1
increased CSF protein and severe demyelination on nerve biopsy are features 5
-
X chromosome-linked CMT (CMTX)
second most common form of CMT overall 5
most commonly due to a mutation in the gap-junction B1 (GJB1) gene 5
Radiographic features
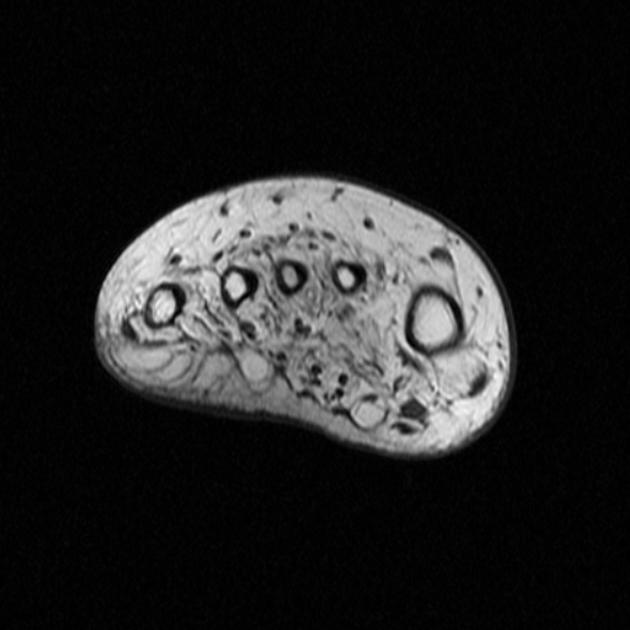
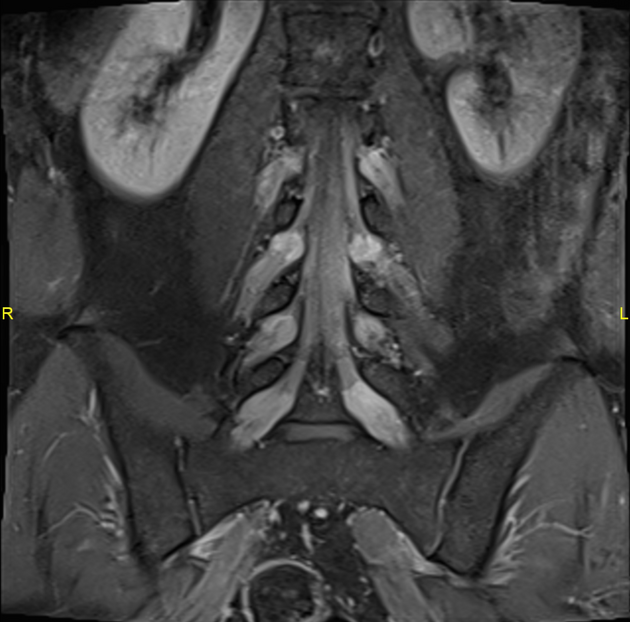
The nerve roots are typically hypertrophic with the onion bulb sign. This represents hypertrophic demyelination. Denervation changes in muscles are apparent.
Some enhancement may be seen on MRI, but it is not usually a prominent feature.
Soft tissue and skeletal abnormalities (e.g. scoliosis) may also be noticeable.
Treatment and prognosis
Unfortunately no effective drug for Charcot-Marie-Tooth disease exists. Treatment is largely supportive with rehabilitation therapy and surgery for skeletal deformities 5. Most patients with CMT1A (the most common form of Charcot-Marie-Tooth disease) will remain able to walk for their entire life. However, the disease has variable severity depending on subtype 5.
History and etymology
This disease is named after French pathologist and neurologist Jean-Martin Charcot (1825-1893), often described as the "father of neurology", French neurologist Pierre Marie (1853-1940), and British neurologist Howard Henry Tooth (1856-1925) 6-8.


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.