
Myofibromas are benign neoplasms of soft tissues of myofibroblastic differentiation.
On this page:
Terminology
The term "myofibroma" is used for solitary lesions and "myofibromatosis" for multicentric lesions.
Epidemiology
Myofibromas can occur at any age, but most predominantly occur in infants and young children, especially before the age of 2 years 1.
Clinical presentation
They can be asymptomatic. If present, symptoms and signs are non-specific 1. Cutaneous myofibromas may present as purplish macules, resembling a vascular neoplasm.
Pathology
Myofibromas can be considered part of a large group of mesenchymal neoplasms called "fibroblastic and myofibroblastic tumors" or "musculoskeletal fibromatoses" that include a broad spectrum of benign, intermediate, and malignant tumors with overlapping histopathology.
The World Health Organization (WHO) classifies myofibromas under the benign category of "pericytic (perivascular) tumors" 4.
Location
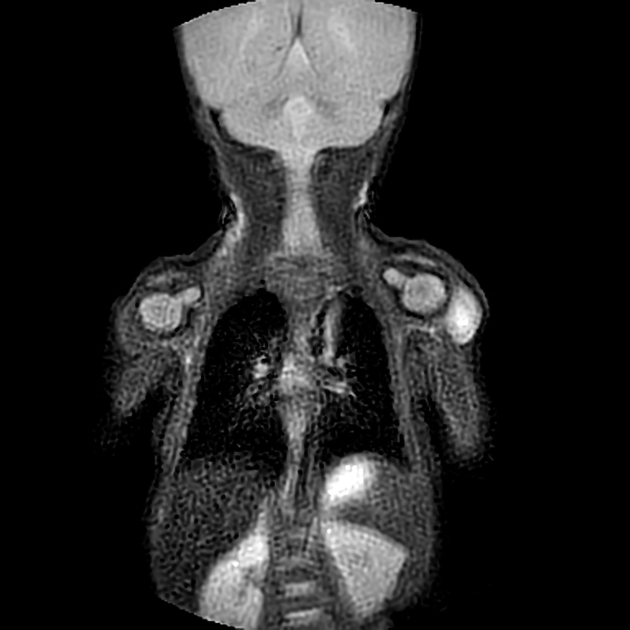
In children, there is a predilection for the head and neck regions > trunk > lower extremities > upper extremities > viscera. Involved organs include the lungs, heart, gastrointestinal tract, liver, kidney, pancreas, or central nervous system.
In adults, myofibromas prefer the dermis, subcutis, or oral cavity, which are more common than deeper lesions in bone, muscle, or aponeurosis. Osseous lesions usually involve craniofacial bones.
Subtypes
There are three main subtypes of myofibromatosis 1:
solitary
multicentric without visceral involvement
multicentric with visceral involvement
Genetics
Most cases are sporadic. However, reports of familial occurrence suggest a possible autosomal dominant or recessive inheritance in some cases of infantile myofibromatosis, which have been linked to mutations in the PDGFR-beta and NOTCH3 genes 2.
Radiographic features
Findings are usually non-specific with a broad differential diagnosis 1.
Plain radiograph
may not be visible
possible dystrophic calcifications
may be lytic if intraosseous
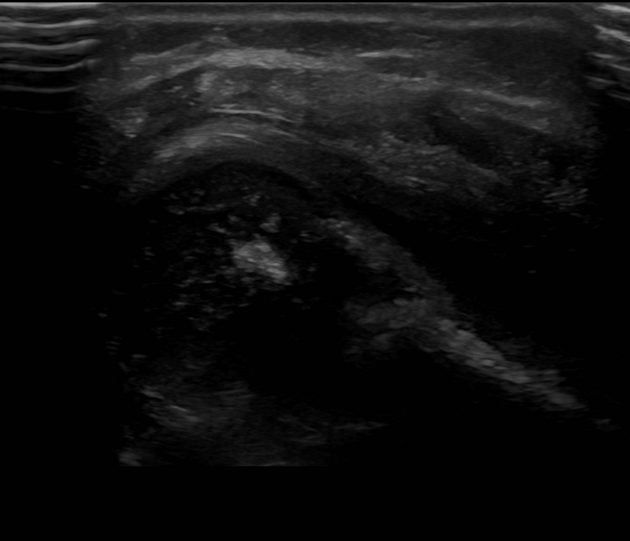
Ultrasound
greyscale: solid hypoechoic mass, central hypo- to anechoic portion due to necrosis
color Doppler: variable echogenicity
CT
isodense to muscle
osseous erosion, if adjacent to bone
possible central necrosis
possible dystrophic calcifications
MRI
ovoid and well-circumscribed
may have a target sign due to central necrosis
Signal characteristics include:
T1: isointense to muscle
T2: hyperintense
T1 C+ (Gd): avid enhancement
Treatment and prognosis
Treatment includes observation or complete local excision. If resected, recurrence rate reported as 7-10% 3. Generalized myofibromatosis involving the viscera can be treated with chemotherapy 1.
History and etymology
Myofibromatosis was initially described by Williams and Schrum in 1951, when it was referred to as "congenital fibrosarcoma" 5. This entity was later renamed "congenital generalized fibromatosis" by Stout in 1954 6, only to be again renamed "infantile fibromatosis" by Chung and Enzinger 3.
Differential diagnosis
Differential diagnosis depends on location. For differential diagnoses of a soft tissue location (such as intramuscular):
metastasis
Practical points
In practice, a well-circumscribed soft tissue lesion with avid enhancement has a very wide differential. For this reason, biopsy is required to make a definitive diagnosis 1. Given how uncommon myofibromas are compared to nerve sheath tumors and other diagnoses, myofibromas will often be very low on the differential.


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.