
Synovial sarcomas are relatively common intermediate-to-high grade malignant soft tissue tumours, often with an initial indolent course, affecting young patients and most commonly involving the soft tissue surrounding the knees.
On this page:
Epidemiology
Synovial sarcomas typically present in adolescents and young adults (15-40 years of age). There may be a mild male predilection (M:F = 1.2:1). They account for 2.5-10% of all soft tissue sarcoma 2-4.
Clinical presentation
The presentation is most often with a slowly enlarging soft tissue mass, which may have been noted for some years and gives a false impression of a benign process 4.
Pathology
Synovial sarcoma is a misnomer because this tumour shows no synovial differentiation but reveals somehow epithelial differentiation, so it stains for epithelial markers (e.g. epithelial membrane antigen and cytokeratin), which synovium does not. Additionally, it has been found in many locations that do not normally have synovium.
The biphasic variant is composed of glandular components (which may be dominant or hidden) and spindle cells. Spindle cells are quite uniform and arranged in fascicular pattern.
Monophasic synovial sarcoma composed of spindle cells and is quite resemble to MPNST, fibrosarcoma, mesenchymal chondrosarcoma and cellular SFT. For establishing a correct diagnosis, an IHC study for keratin and EMA is very helpful. Nearly 90% of monophasic variants are at least focally positive for keratin or EMA.
SS18-SSX fusion antibody is 95% sensitive and specific for synovial sarcoma.10
Location
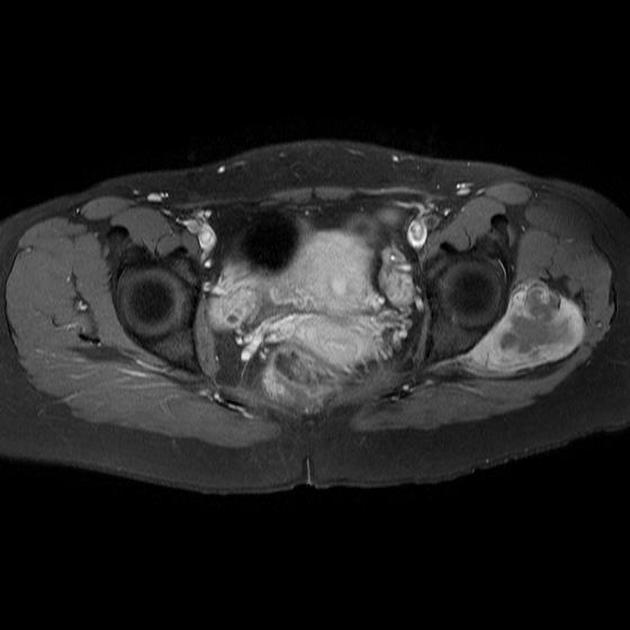
The most common location for these tumours is within the deep soft tissues adjacent to large joints, e.g. knee and popliteal fossa. While these tumours arise near joints, it is rare for them to arise from the joint itself and despite their name, they do not arise from synovial structures, e.g. joints, tendon sheaths and bursae.
-
extremities: 80-95% 1
lower limb: 60-70%
upper limb: 15-25%
from soft tissues: 90-95%
from joint: 5-10%
-
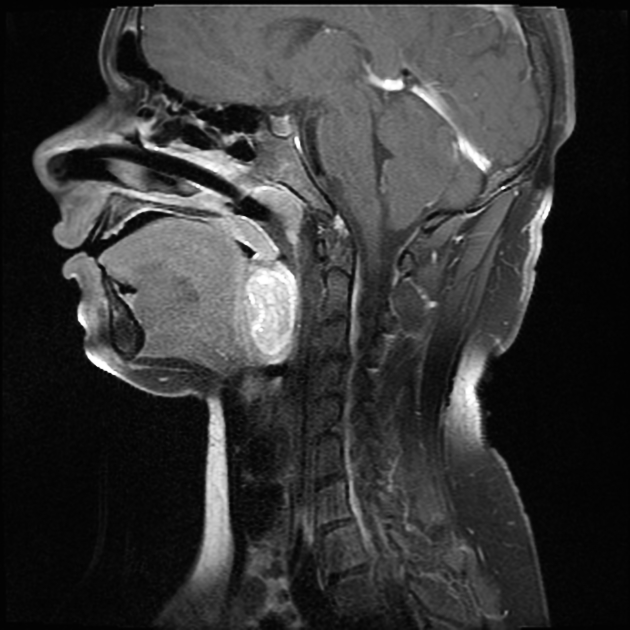
head and neck: 5%
tongue: rare 9 (synovial sarcoma of the tongue)
conjunctiva 3
CNS
chest wall: rare 6
-
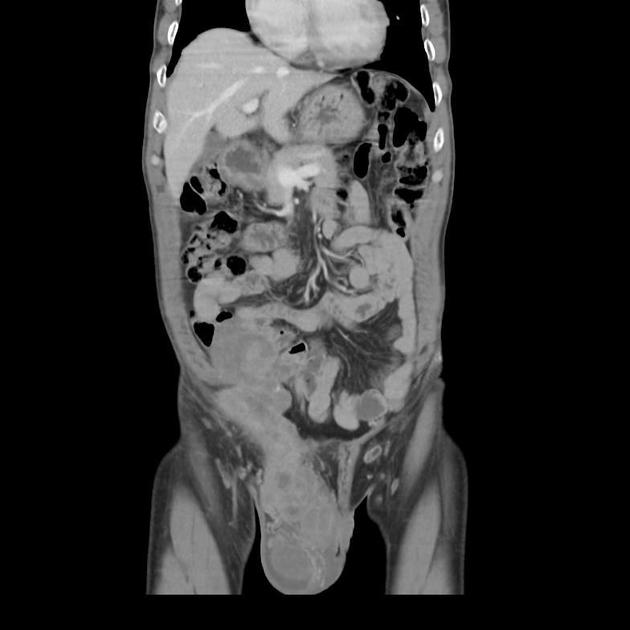
viscera: rare
lung 2, kidney, prostate, oesophagus, heart, pericardium, pleura
Macroscopic appearance
Macroscopically, they have non-specific appearances. They are well or poorly-defined heterogeneous masses with common areas of haemorrhage and necrosis.
Microscopic appearance
They are divided histologically into four subtypes:
biphasic: 20-30%
monophasic fibrous: 50-60%
monophasic epithelial: very rare
poorly differentiated: 15-25%
Genetics
Cytogenetic aberration of the t(X;18) translocation is highly specific, seen in over 90% of cases 1-4.
Radiographic features
A soft tissue mass near but not in a joint in a young patient (15-40 years old), particularly if dystrophic calcification is present, is very suggestive.
Plain radiograph
Radiographs may be normal unless the mass is large or contains dystrophic calcifications found in up to 30% of cases 1. These calcifications are non-specific within the lesion's periphery and are usually not osteoid or chondroid in appearance.
Occasionally, smooth pressure erosions may be visible on the underlying bone, which should not be mistaken for a sign of a benign process.
Alternatively, direct bony invasion is seen in ~30% (range 11-50%) of cases, with aggressive bone features (permeative, geographic, or moth-eaten appearances) 4.
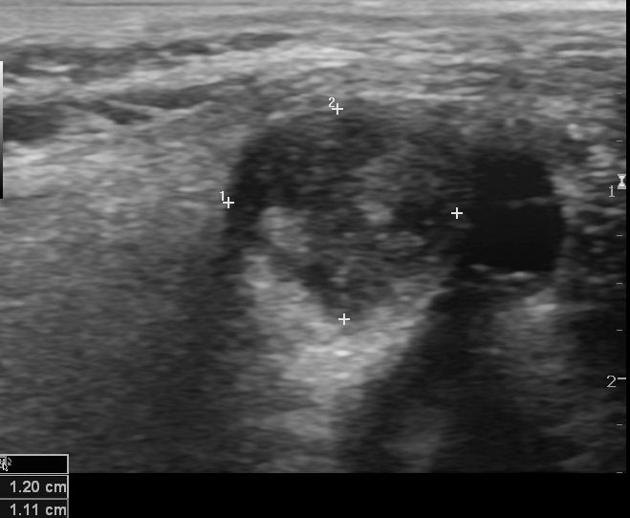
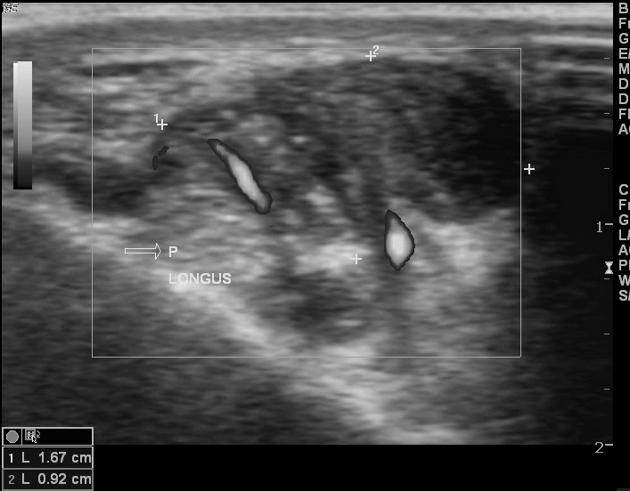
Ultrasound
Non-specific findings, with a heterogeneous predominantly hypoechoic mass. Intra-lesion flow is usually associated with more aggressive lesions.
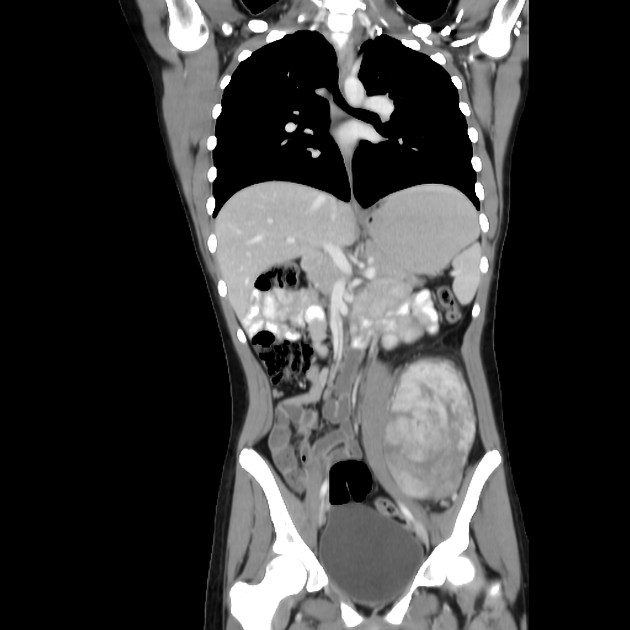
CT
CT is non-specific, similar to ultrasound. It appears as a soft tissue mass of heterogeneous density and enhancement. CT is, of course, more sensitive to calcifications than either radiographs or MRI.
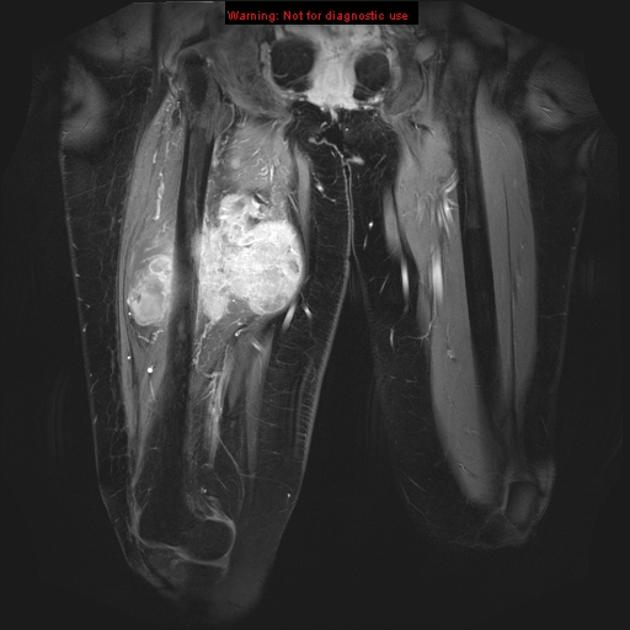
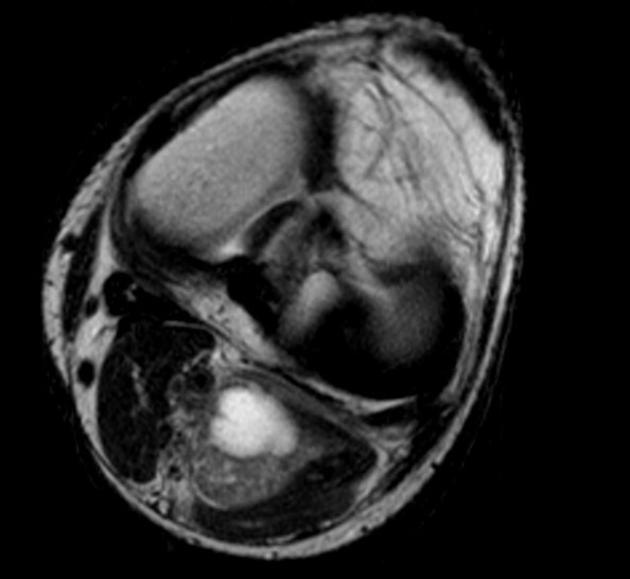
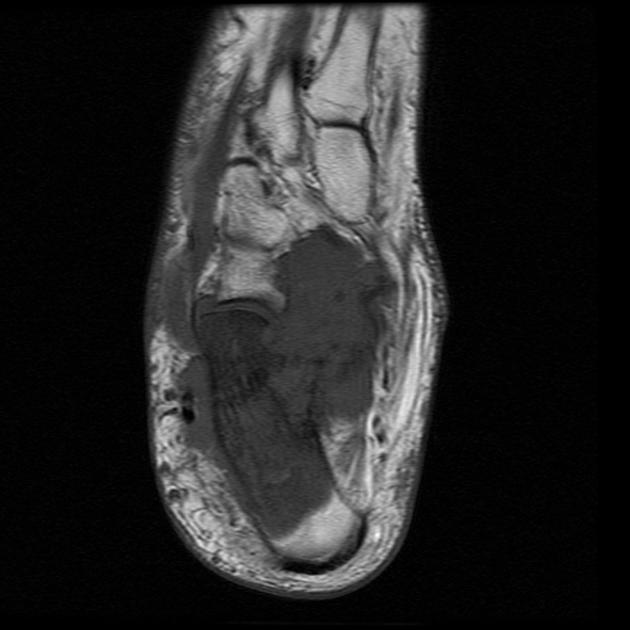
MRI
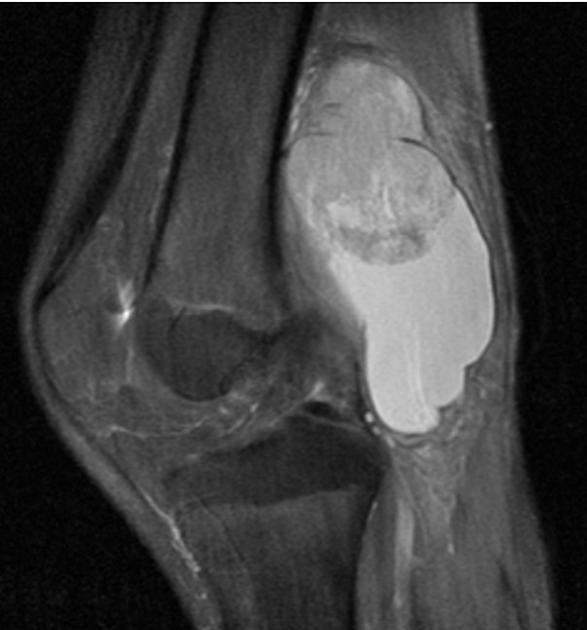
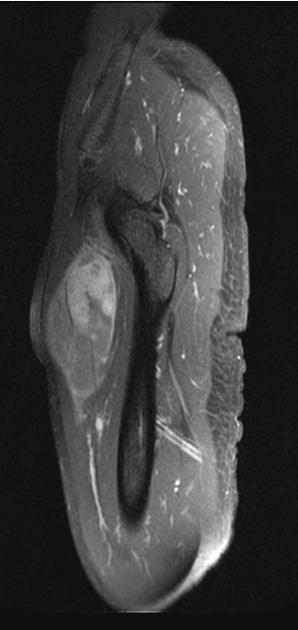
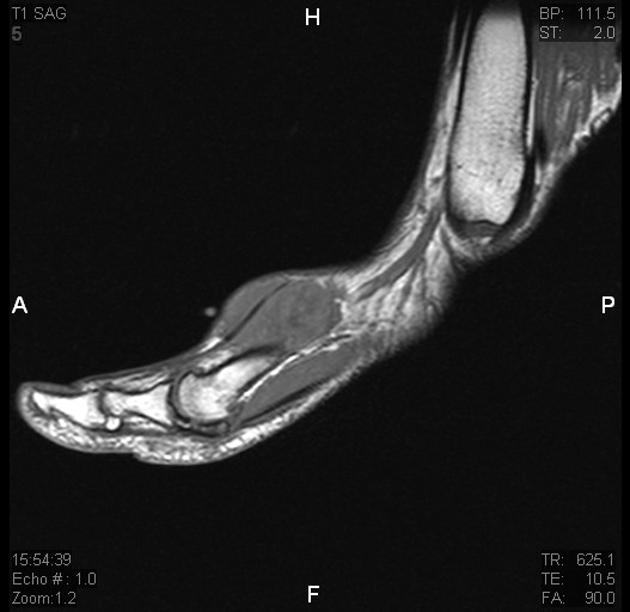
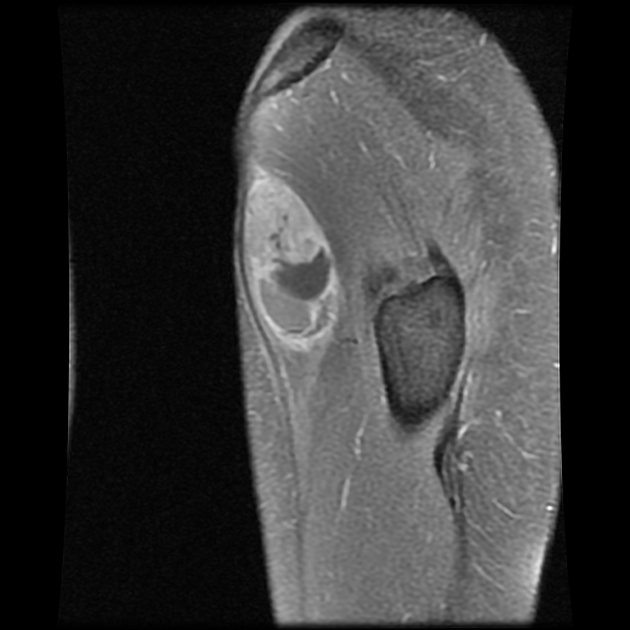
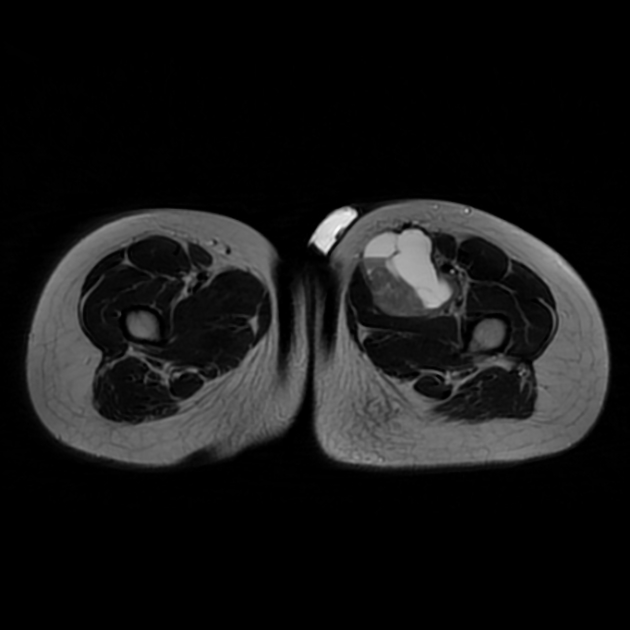
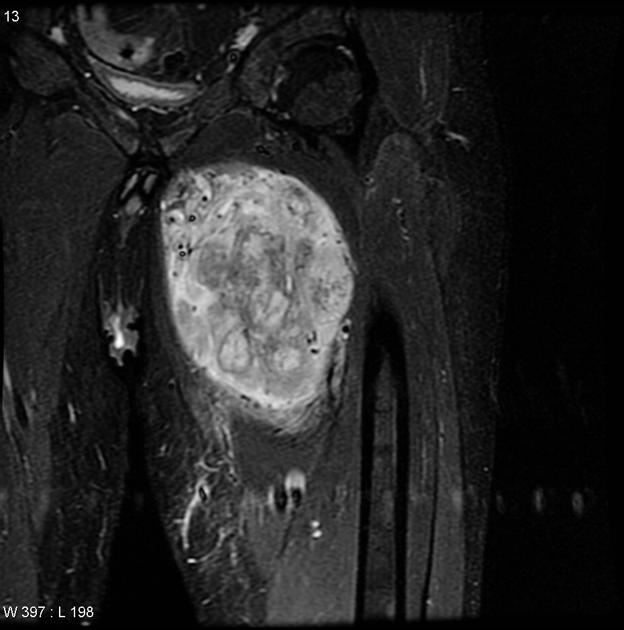
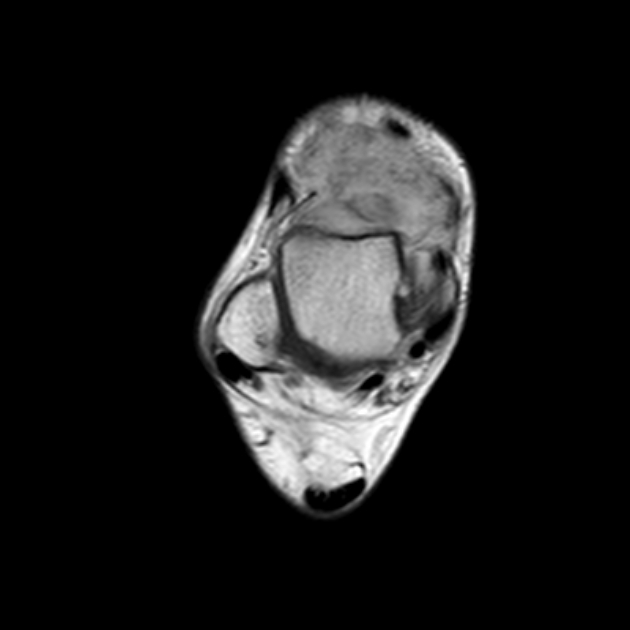
MRI is the modality of choice to stage the tumour locally, although, again, imaging findings are not pathognomonic. The mass is usually large and variably well-defined (smaller lesions tend to be better circumscribed).
-
T1
iso- (slightly hyper-) intense to muscle
heterogeneous
-
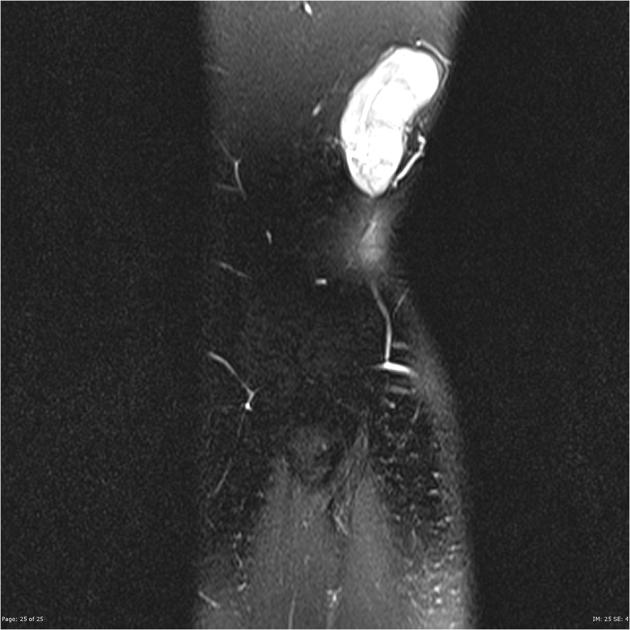
T2
mostly hyperintense
the markedly heterogeneous appearance of synovial cell sarcomas on fluid-sensitive sequences results in so-called "triple sign" which is due to areas of necrosis and cystic degeneration with very high signal, relatively high signal soft tissue components and areas of low signal intensity due to dystrophic calcifications and fibrotic bands
due to the high tendency of lesions to bleed, there might be areas of fluid-fluid levels known as "bowl of grapes" are seen in up to 10-25% of cases
-
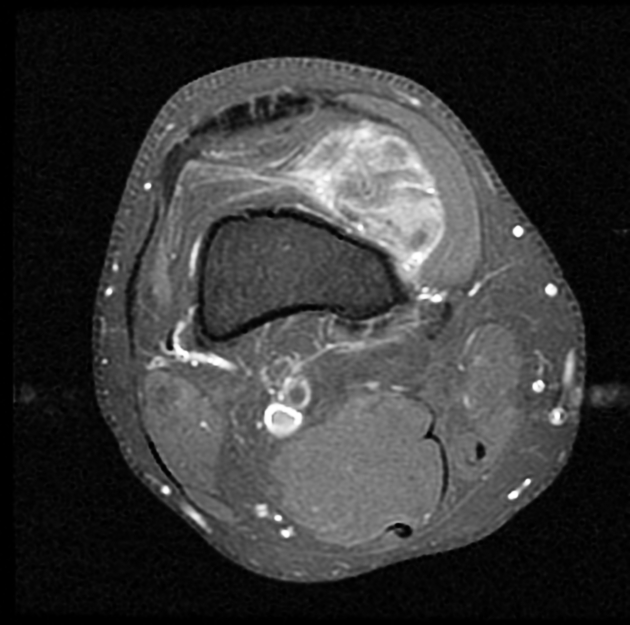
T1 C+ (Gd)
enhancement is usually prominent and can be diffuse (40%), heterogeneous (40%), or peripheral (20%) 1
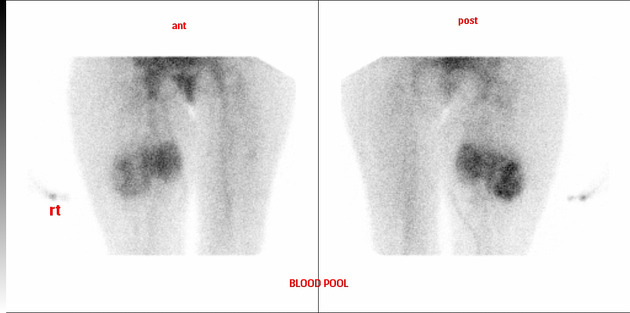
Nuclear medicine
On bone scans, synovial sarcomas typically have heterogeneously increased dynamic and blood pool uptake, with some increase in delayed images, but usually less intense.
Angiography (DSA)
This imaging test is not usually performed. These tumours are usually hypovascular 1.
Treatment and prognosis
Treatment involves a combination of surgery and usually adjuvant radiotherapy +/- chemotherapy. Radiotherapy is particularly useful in treating tumours where an adequate clear margin cannot be achieved, and ideally, radiotherapy is administered pre-operatively.
Good prognostic variables include:
small size
located in the extremity
younger age <20 years of age
solid homogeneous mass
presence of calcification
biphasic histology (controversial)
Poor prognostic variables include:
large size (>5 cm): the most important factor
located in the trunk or head and neck
older patients
cystic/haemorrhagic components
marked heterogeneity
-
histology
poorly differentiated histology
rhabdoid cells
extensive tumour necrosis
high nuclear grade
p53 mutations
high mitotic rate (>10 mitoses per 10 high-power field)
Overall 5-year survival is between 36-76%. Both local recurrence (30-50%) and distant metastases are frequent (40-70%), most commonly to the lungs as cannonball metastases (~80%), bones (~15%), regional lymph nodes (~10%), followed by chest wall/abdomen (~7.5%) 7,8.
Differential diagnosis
General imaging differential considerations include:





 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.