
Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP), also known as hereditary diffuse leukoencephalopathy with spheroids (HDLS) and pigmentary orthochromatic leukodystrophy (POLD), refers to a rare inherited autosomal dominant disease characterized by an adult-onset leukodystrophy that usually leads to death in around 5-7 years. It is considered to belong to the microgliopathies.
On this page:
Terminology
For many years hereditary diffuse leukoencephalopathy with spheroids (HDLS) and pigmentary orthochromatic leukodystrophy (POLD) were considered to be two separate hereditary leukoencephalopathies. Sometimes HDLS was also called neuroaxonal leukodystrophy. The striking similarities in clinical presentation and histology suggested a link between the two diseases for a long time and contemporary literature considers HDLS and POLD to be part of the same disease spectrum, which researchers have recommended calling adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) 1.
Epidemiology
Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia is considered a rare disease, typically manifesting between ages 30 and 50 years. Its exact prevalence is unknown, as it has been previously mistaken for many other diseases and it might thus continue to be underdiagnosed.
While it is usually an inherited disease, there exist case reports of proven de novo mutations and mutation carriers without clinical symptoms over the age of 70 years 2.
Clinical presentation
Patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia can have a wide variety of symptoms that usually exhibit a rapid progression and lead to death within a couple of years due to progressive motor impairment.
Typical symptoms are:
depression, which might precede the other symptoms
neuropsychiatric symptoms, progressing to dementia
motor impairment, with extrapyramidal and pyramidal symptoms that usually lead to tetraparesis
Pathology
Genetics
Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia is caused by autosomal dominantly inherited mutations in the colony-stimulating factor 1 receptor (CSF1R) gene. The mutations can be proven by genetic testing of blood samples.
Microscopic appearance
Before the identification of the underlying genetic cause of the disease, a brain biopsy was needed to establish the diagnosis. Neuropathologic findings usually show gliosis, white matter destruction with pigmented glia and/or axonal spheroids and macrophages.
Radiographic features
CT
Small periventricular calcifications, often only seen on thin sagittal reconstructions with a bone kernel are a typical imaging finding 11. Affected areas show a low attenuation and no contrast enhancement.
MRI
Presymptomatic mutation carriers
Mutation carriers usually show non-specific T2 hyperintense lesions of the white matter that are pronounced for the age of the patient. MR spectroscopy in these lesions is usually normal without characteristic findings.
Symptomatic patients
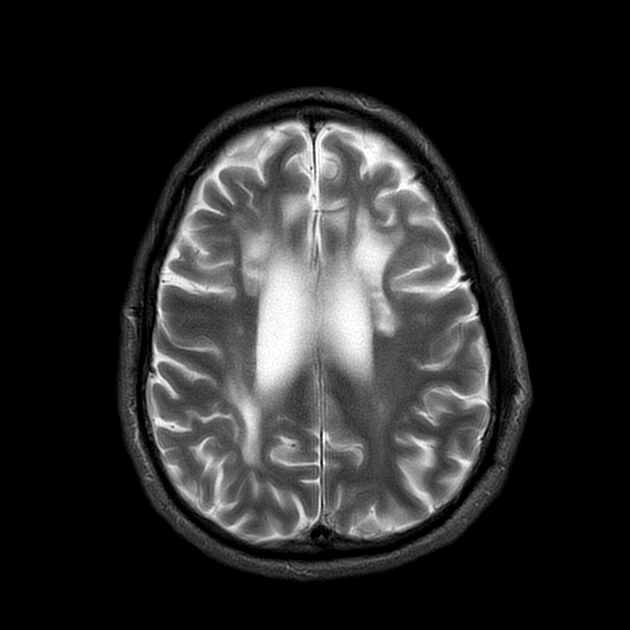
Characterized by bilateral, asymmetric patchy or confluent areas of subcortical and deep white matter signal change, usually most pronounced in the frontal lobe followed by the parietal lobe with a relative sparing of the temporal lobe. The corticospinal tract is usually affected late in the disease course. Progression leads to a severe atrophy of the supratentorial white matter. The cerebellum and brainstem are usually spared 3,4.
T1: affected areas are low in signal
T1 C+ (Gd): no enhancement is visible
T2/FLAIR: hyperintense
DWI: small spots of diffusion restriction, that can be visible over months, is a characteristic finding 3
-
MR spectroscopy: (in symptomatic patients)
reduced N-acetylaspartate (NAA)
increased choline
increased myo-inositol
increased lactate
Treatment and prognosis
There exists no accepted specific therapy and the disease is inevitably fatal within a couple of years. In a few cases, hematopoietic stem cell transplantation was attempted as an individual cure, leading to clinical stabilization in some cases but also therapy-associated death in one case 10.
Differential diagnosis
The main differential for adult-onset leukoencephalopathy with axonal spheroids and pigmented glia is leukoencephalopathy due to autosomal recessive mutations in the mitochondrial alanyl-transfer RNA (tRNA) synthetase gene (AARS2-L), another adult-onset leukodystrophy with similar pathologic findings. The clinical presentation and imaging findings in AARS2-L strongly resemble those of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia, but there are subtle differences 8,9.
General imaging differential considerations include 2-6:
Clinical differential considerations include 2-6:



 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}