
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL)
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) is a systemic genetic disorder affecting the cerebral small vessels, spine and hair follicles.
CARASIL is an HTRA1-related cerebral small vessel disease, and thus is closely related to, but distinct from, heterozygous HTRA1-related cerebral small vessel disease.
On this page:
Terminology
It should not be confused with its autosomal dominant counterpart, CADASIL. Autosomal recessive inherited diseases are usually more severe than autosomal dominant genetic disorders and CARASIL is no exception to this general rule.
Epidemiology
CARASIL is a very rare disease with roughly 50 cases reported, mostly from inside Asia, although it has also been reported in a European family 1,2.
The true prevalence is unknown, and CARASIL is probably under-recognized since no founder haplotype has been identified, and the encountered European disease carriers support the theory of a wider distribution 1,2,4.
Clinical presentation
Typically patients present in young adulthood (twenties and thirties) with:
premature alopecia (as early as teenage years; most common initial symptom)
relapsing ischemic stroke-like episodes (typically in the 3rd decade) with gradually progressive vascular dementia
not infrequently, symptomatic epilepsy (secondary to infarctions)
relapsing severe low back pain and deforming spondylosis (present in roughly 80% of patients, notably early onset)
Pathology
Macroscopic appearance
Shows cerebral arteriosclerotic changes, most intense in white matter and basal ganglia.
Microscopic appearance
Intense arteriosclerosis is seen predominantly in the small penetrating arteries. Aids in differentiation from other entities are the lack of amyloid deposition and absence of the granular appearance characteristics of CADASIL.
Extraneural changes are less severe and although they include the cutis, skin biopsies are not helpful in establishing the diagnosis.
Genetics
CARASIL is, after CADASIL, the second known genetic form of ischemic CNS disease caused by non-hypertensive microangiopathy in which the causative gene defect has been identified.
Homozygous or compound heterozygous mutations in the HTRA1 gene on chromosome 10q (10q25.3-q26.2) are responsible for CARASIL. It codes for the ubiquitous HTRA1 enzyme, which regulates signaling by proteins in the transforming growth factor-beta (TGF-β) family, which again is essential for multiple critical cell functions including angiogenesis. The pivotal role of TGF-β in CARASIL is hypothesized, but not totally clarified.
Radiographic features
CNS manifestations
CT
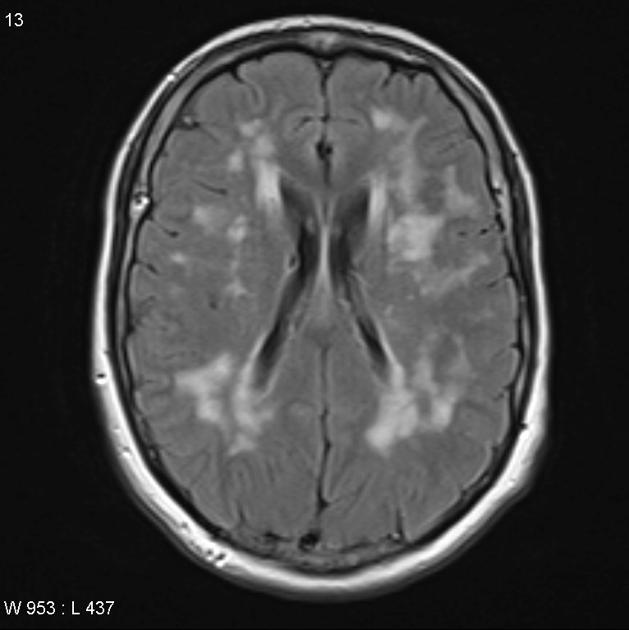
diffuse homogeneous leukoaraiosis
ex-vacuo ventriculomegaly, predominantly affecting the lateral ventricles
in ~25% of patients, small hypodense areas may be seen located in the pontine base (possibly presenting Wallerian degeneration of the corticospinal tract)
MRI
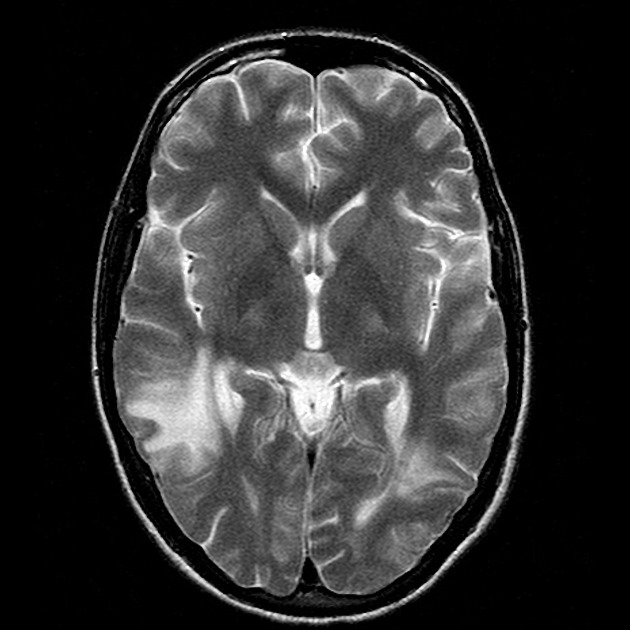
diffuse leukoencephalopathy
-
multiple lacunar infarctions in the deep nuclei, most often
high T2 signal connecting the middle cerebellar peduncles across the pons; arc sign 8
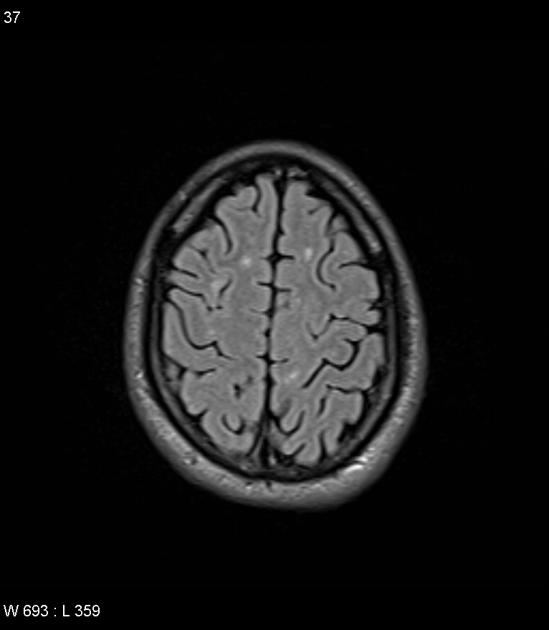
cerebral microhemorrhages may be seen 8
Musculoskeletal manifestations
Changes may be appreciated by plain radiograph, CT or MRI, and are those of:
-
deforming spondylosis with or without intervertebral space narrowing, located in the cervical and/or thoracolumbar spine
typical location upper lumbar regions (which is in some contradiction to the sporadic spondylosis)
elbow or knee osteoarthritis
Treatment and prognosis
As there are no causative treatment options, treatment focuses on the prevention of ischemic strokes. Interestingly, treatment with known antiplatelet agents and anticoagulant lacks evidence.
Another goal of management is to prevent the onset of dementia symptoms. Genetic counseling is also an important part of primary care.
History and etymology
It was first described by S Nemoto, a Japanese physician, in 1960 where it was initially described as part of the Binswanger spectrum 5,6. Nemoto went on to publish a seminal case series with S Maeda et al. in 1976 7. The disease is sometimes known as Nemoto disease or Maeda syndrome in recognition of their contributions.
Differential diagnosis
-
symptom onset in CARASIL is 10-15 years earlier
migraine and depression are not seen in CARASIL
early white matter changes in CARASIL are homogenous rather than punctuate as seen in CADASIL
cathepsin A-related arteriopathy with strokes and leukoencephalopathy (CARASAL)
other cerebral small vessel diseases (SVD)
-
progressive multiple sclerosis
involvement of subcortical U-fibers is less pronounced in CARASIL


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.