
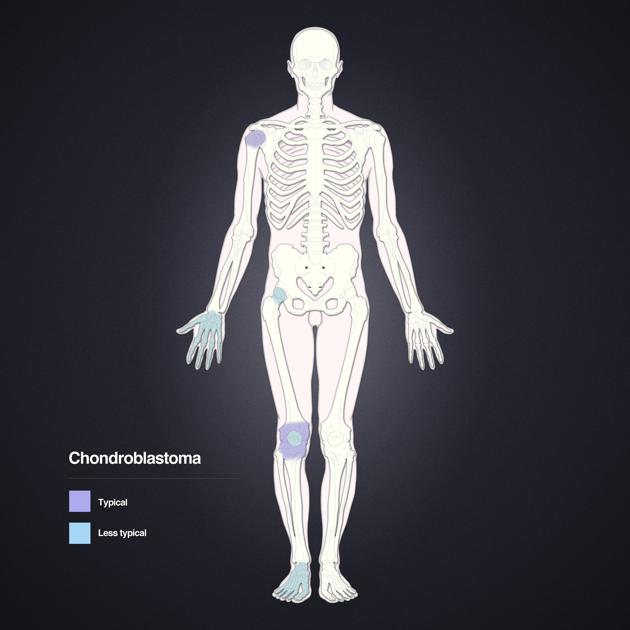
Chondroblastomas are benign chondrogenic bone neoplasms characteristically arising in the epiphysis or apophysis of a long bone in young patients. Despite being rare, they are one of the most frequently encountered benign epiphyseal neoplasms in skeletally immature patients 1.
Chondroblastoma accounts for one of the 'C's in the popular mnemonic for lucent bone lesions FEGNOMASHIC.
On this page:
Epidemiology
Chondroblastomas represent <1% of all primary bone tumours 1,2 occurring predominantly in the immature skeleton of young patients in the second and early third decade (10-25 years) with an overall male predilection 1-3.
Diagnosis
The diagnosis of chondroblastomas is based on a combination of typical radiological and pathological features 1.
Diagnostic criteria
Diagnostic criteria according to the WHO classification of soft tissue and bone tumours (5th edition) 1:
Essential criteria are 1:
osteolytic bone tumour in an epiphyseal or apophyseal location
sheets of chondroblasts with interspersed osteoclastic giant cells and eosinophilic chondroid matrix
The following criteria are desirable 1:
network of pericellular chickenwire-calcifications
presence of H3.3 mutation either by H3-3A/H3-3B analysis or by p.Lys.36Met (K36M) expression
Clinical presentation
Clinical symptoms vary with the location of the tumour and include bone pain, local tenderness, joint effusion with stiffness and restricted motion or muscle atrophy 1-3.
Complications
Complications include 1-3:
joint effusion
tumour recurrence
benign lung metastases (in exceptional cases)
Pathology
Chondroblastomas are well-defined tumours composed of chondroblastic cells, islands of a chondroid matrix with a sclerotic rim. They usually show an eccentric position in the epiphyses and apophyses of long bones 1.
Aetiology
The aetiology of chondroblastomas is not known 1.
Location
Most chondroblastomas occur in the subchondral epiphyseal region of long bones such as the proximal or distal femur, the proximal tibia and the proximal humerus 1-3. They can also occur in the talus, calcaneus, patella and pelvic bones e.g. the acetabulum and less frequently affect the ribs the spine or small bones of hands and feet 1 or the craniofacial skeleton 2,3. They are usually confined to a single bone.
Macroscopic appearance
Grossly chondroblastomas display the following features 1-3:
greyish-yellow appearance with haemorrhage
small calcifications often leading to a gritty and chalky cut surface
sharply demarcated from adjacent bone
aneurysmal bone cyst-like changes or areas of necrosis might be present
Microscopic appearance
Histologically, chondroblastomas are characterised by the following 1:
sheets of intermediate-sized chondroblastic ovoid to polygonal cells with eosinophilic cytoplasm
centrally placed nuclei often with longitudinal nuclear grooves (“coffee bean” nuclei)
possible cellular atypia and occasional mitoses
interspersed osteoclastic giant cells
islands of chondroid matrix
pericellular lace-like "chicken-wire calcification" (pathognomonic)
commonly aneurysmal bone cyst-like changes
The presence of occasional giant multinucleated cells may lead to the incorrect diagnosis of a giant cell tumour.
Immunophenotype
On immunohistochemistry diffuse nuclear expression of p.Lys36Met (K36M), an antibody against H3.3B is characteristic 1.
Genetics
Chondroblastomas are characterised by a p.Lys36Met substitution on the H3-3B (H3F3B) or less frequently H3-3A (H3F3A) genes 1.
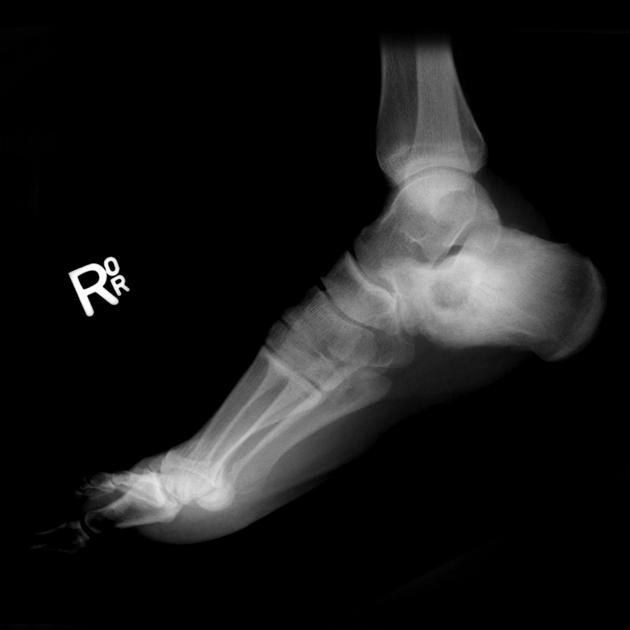
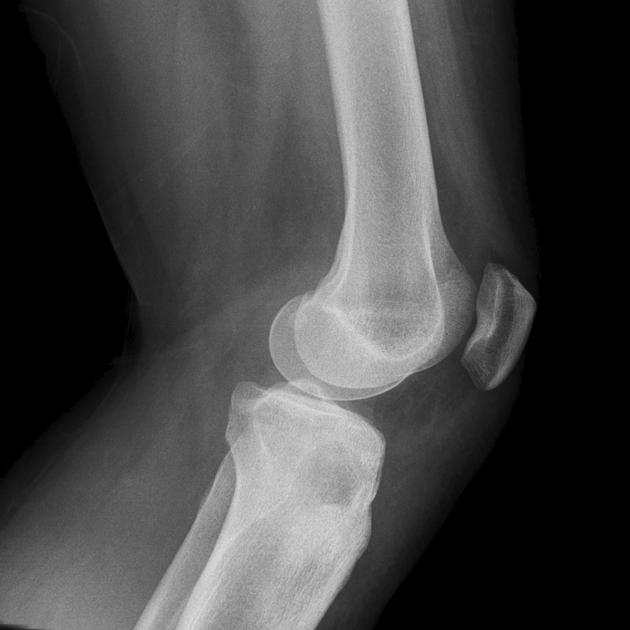
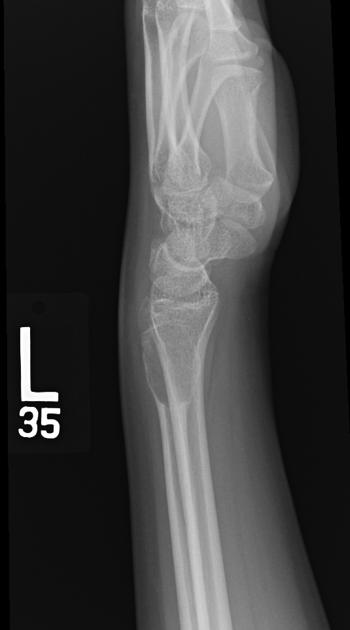
Radiographic features
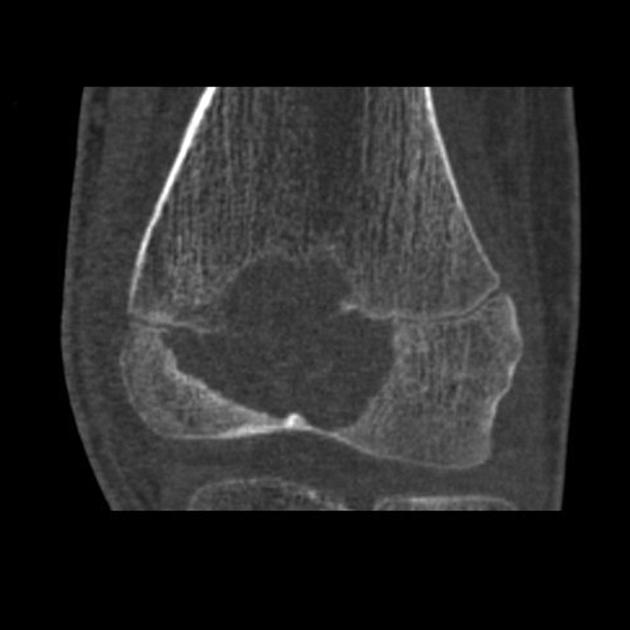
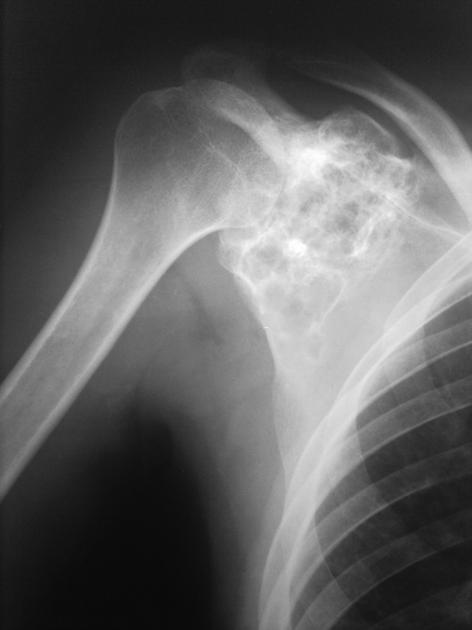
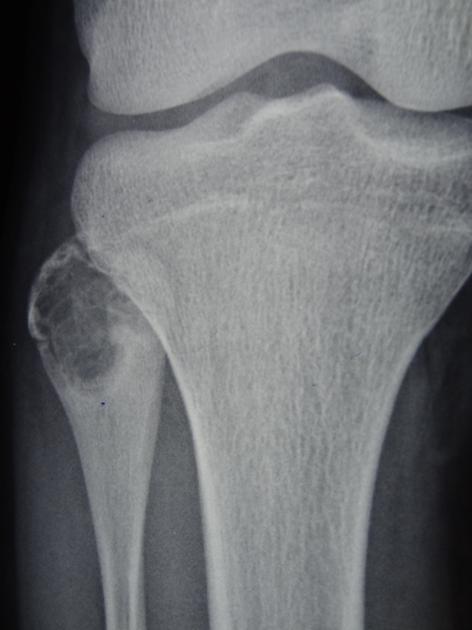
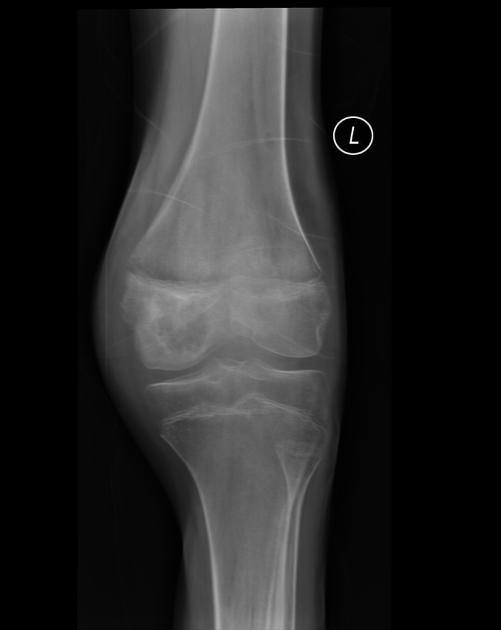
Chondroblastomas are well-defined lytic bone lesions with geographical bone destruction and thin sclerotic margins. They are usually eccentric epiphyseal or apophyseal located adjacent to the growth plates 1-3. As they grow larger they can extend into the metaphyses. They might be associated with joint effusion. Lesions are usually <5 cm on detection 1,3.
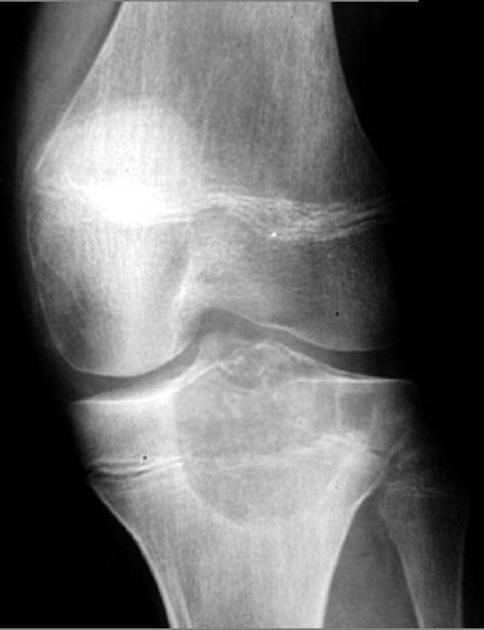
Plain radiograph
On plain radiographs, chondroblastomas display the following characteristics 1,2:
radiolucent bone lesion
internal “fluffy” calcifications (in about half of the cases)
narrow zone of transition
bone mineralisation, trabeculation
endosteal scalloping, cortical thinning, cortical erosion
cortical breaches with soft tissue invasion (rarely)
periosteal reaction in a metadiaphyseal location 4
joint effusion (in up to one-third of patients)
Among epiphyseal lesions, the presence of solid or layered periosteal reaction distant to the lesion (involving the diaphysis) is distinctive 7.
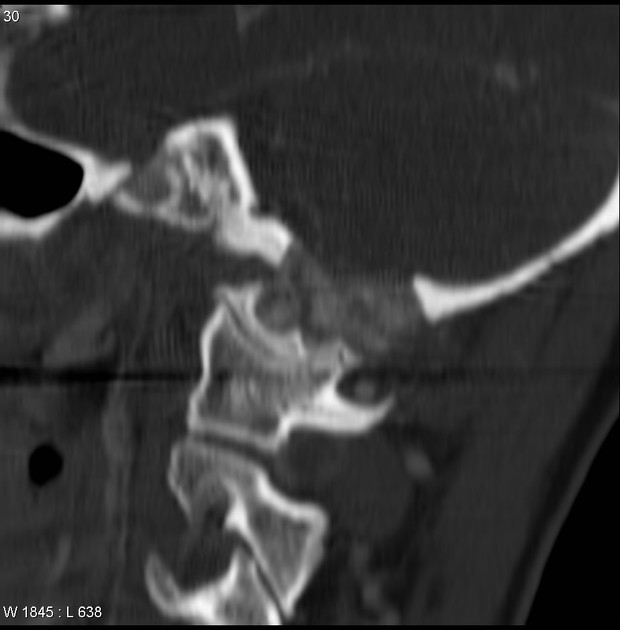
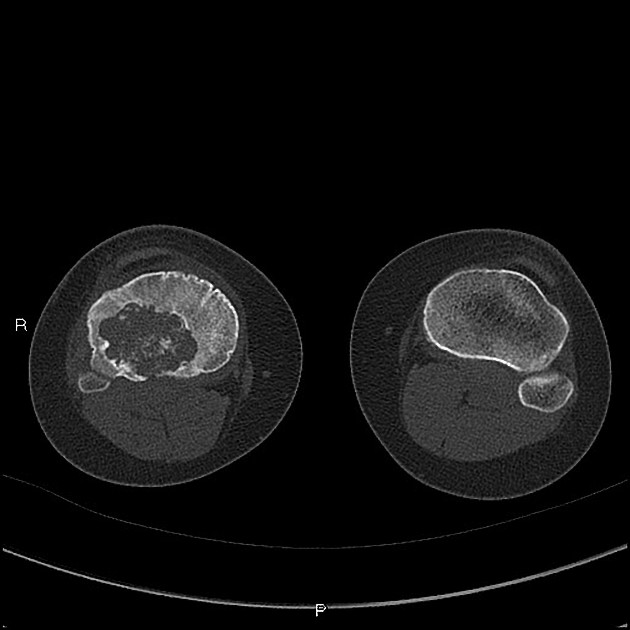
CT
Chondroblastomas are solitary lucent bone lesions.
Features like internal calcifications, cortical involvement and cortical destruction can be more readily assessed than on plain radiography 7,8.
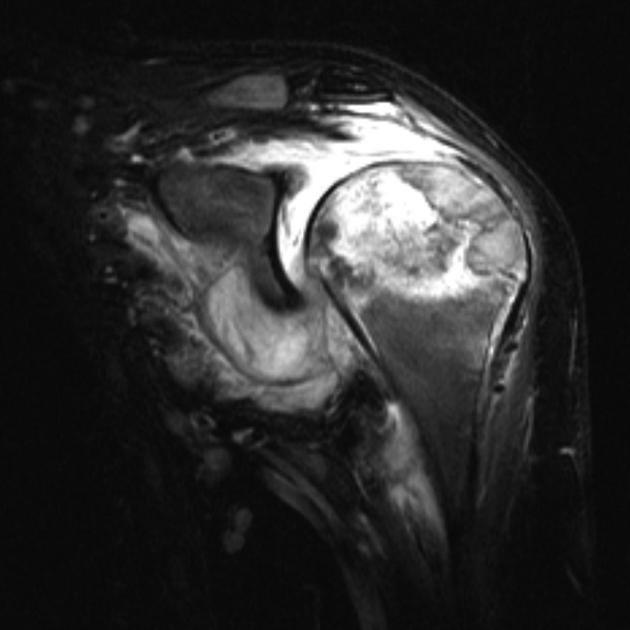
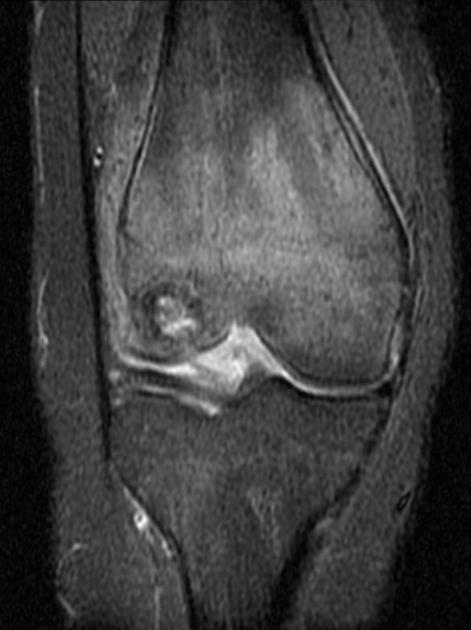
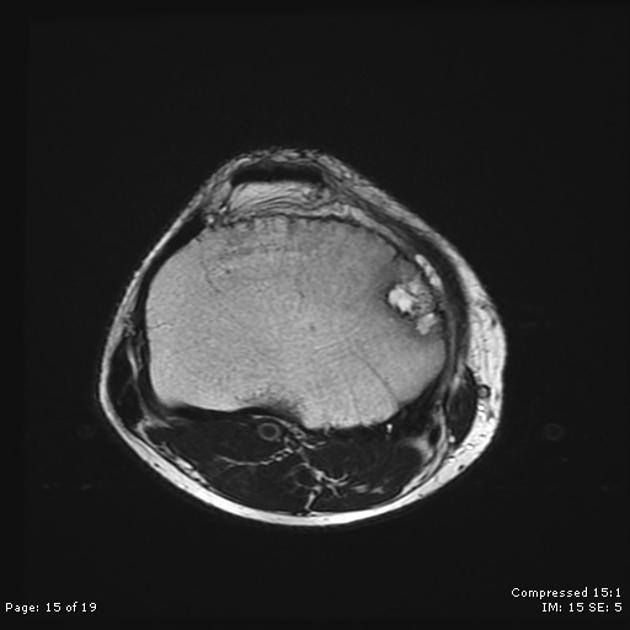
MRI
On MRI chondroblastomas usually demonstrate the following features 6-9:
lobular internal architecture
a thin hypointense sclerotic rim on T1 and T2 weighted images
surrounding bone marrow oedema
cortical involvement
soft tissue oedema
possible fluid-fluid levels in the presence of aneurysmal bone cyst-like changes
Signal characteristics
T1: intermediate signal
T2: variable, heterogeneous intermediate signal intensity
STIR: high signal
T1 C+ (Gd): heterogeneous moderate enhancement together with enhancement of surrounding bone and soft-tissue oedema
Nuclear medicine
Bone scintigraphy
Chondroblastomas usually demonstrate increased radionuclide uptake 3 in the epiphysis and metaphysis, especially in the uninvolved bone adjacent to the chondroblastoma 7.
Radiology report
The radiological report should include a description of the following 10:
location and size
tumour margins and transition zone
relation to the growth plate and articular surface
cystic and solid tumour components
fluid-fluid levels suggesting aneurysmal bone cyst-like changes
-
additional features
cortical involvement
soft tissue extension
pathologic fracture
aggressive periosteal reaction
surrounding bone marrow oedema
solid mass-like enhancement
associated joint effusion
On CT and MRI the lesion can be categorised as Bone-RADS 4 unless histology has been already obtained 10.
Treatment and prognosis
Chondroblastomas are benign tumours. Treatment typically consists of curettage and packing of the resulting cavity with or without adjuvant therapy 1-3,11-13. Radiofrequency ablation may be used 1,8,11.
Local recurrence is site-specific, higher in flat bones and can happen in up to 18% usually in an interval ranging from 6 months to 8 years 1,2.
Treatment may be also complicated by growth plate injury, which can lead to growth arrest and limb length discrepancy 8.
Benign lung metastases are exceptionally rare 1-3.
History and etymology
A chondroblastoma has been described as a cartilage-containing tumour by Kolodny in 1927 3,4 and as a “calcifying giant cell tumour” by Ewing in 1928 2,4.
In 1931 this lesion was described by the American physician Ernest Armory Codman (1869-1940) as an ‘epiphyseal chondromatous giant cell tumour of the proximal humerus’ of whom it adopted the name Codman tumour 2,3,14.
In 1942 the American pathologists Henry Louis Jaffe (1896-1979) and Louis Lichtenstein (1906-1977) designated this tumour as a benign chondroblastoma of bone 15.
Differential diagnosis
The differential is that of other lesions with a predilection for the epiphysis or apophysis (see differential for an epiphyseal lesion). Specific lesions to be considered include 2,3,16:
clear cell chondrosarcoma: see chondroblastoma vs clear cell chondrosarcoma
osteomyelitis with abscess, e.g. Brodie abscess
giant cell tumour: older age group (closed physis)
The presence of bone marrow oedema frequently seen surrounding chondroblastomas is helpful, as it is not a standard feature of chondromyxoid fibromas, giant cell tumours or enchondromas 8.



 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}