
Medullary cystic disease complex belongs to group of pediatric cystic renal diseases characterized by progressive tubular atrophy with glomerulosclerosis (chronic tubulointerstitial nephritis) and multiple small medullary cysts.
On this page:
Epidemiology
There is no recognized gender predilection.
Clinical presentation
Presentation with polydipsia and polyuria, due to initial tubular injury, tending to progress to end stage renal failure, growth delay or restriction, lethargy.
Three clinical variants based on age of onset for end stage renal disease (ESRD):
infantile: before 2 years of age
juvenile (a.k.a. nephronophthisis): most common form, age of onset 10
adolescent (a.k.a. medullary cystic kidney disease): usually develops in patients in their thirties
There can be a clinical triad comprising of uremia, anemia, and salt wasting (hyponatremia, hypokalemia).
Pathology
It comprises a group of related conditions characterized by multiple cysts typically at the corticomedullary junction and medulla. The medullary cysts are small. There can be associated atrophy and fibrosis of the basement membrane of the proximal and distal tubules which leads into interstitial fibrosis and end stage renal disease.
Variants
familial nephronophthisis: autosomal recessive (40%)
sporadic: non-familial (20%)
retinal renal syndrome: autosomal recessive (15%) associated with retinitis pigmentosa
adult onset medullary cystic disease: autosomal dominant (15%)
Radiographic features
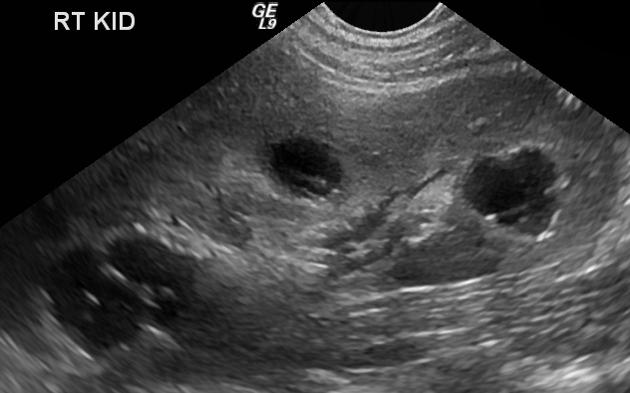
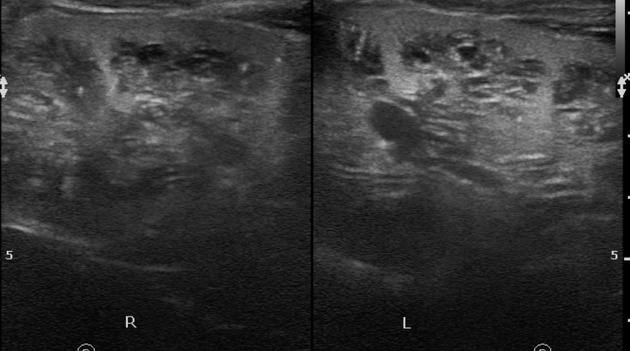
Normal to small kidneys with multiple small (<1.5 cm) medullary cysts (sometimes cysts are too small to visualize) at the corticomedullary junction.


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.