Osteogenesis imperfecta (OI) refers to a heterogeneous group of congenital, non-sex-linked, genetic disorders of collagen type I production, involving connective tissues and bones.
The hallmark feature of osteogenesis imperfecta is osteoporosis and fragile bones that fracture easily, as well as, blue sclera, dental fragility and hearing loss. There is extreme variation in clinical symptoms based on genetic basis and subtypes. Osteogenesis imperfecta affects both bone quality and quantity (i.e. bone mass).
On this page:
Epidemiology
The estimated incidence is approximately 1 in every 12,000-15,000 births 2. Osteogenesis imperfecta occurs with equal frequency among males and females and across races and ethnic groups. The lifespan varies with the type (see osteogenesis imperfecta classification).
Associations
Clinical presentation
The clinical presentation of osteogenesis imperfecta is highly variable, ranging from a mild form with no deformity, normal stature and few fractures to a form that is lethal during the perinatal period.
In general, four major clinical features characterize osteogenesis imperfecta 5:
osteoporosis with abnormal bone fragility
blue sclera
dentinogenesis imperfecta
hearing impairment
Other features include ligamentous laxity and hypermobility of joints, short stature and easy bruising.
Pathology
A fundamental pathology in osteogenesis imperfecta is a disturbance in the synthesis of type I collagen, which is the predominant protein of the extracellular matrix of most tissues. In bone, this defect results in osteoporosis, thus increasing the tendency to fracture. Besides bone, type I collagen is also a major constituent of dentin, sclerae, ligaments, blood vessels and skin 4.
Genetics
Osteogenesis imperfecta is the result of a mutation in one of the two genes that carry instructions for making type 1 collagen. Mutations in the COL1A1 and COL1A2 genes, which encode the α1 and α2 polypeptide chains 7, are responsible for >90% of all cases.
Depending on the type, the inheritance of the disorder can be autosomal dominant (>95%), autosomal recessive (<10%) or by sporadic mutation 2,6,7.
Classification
The first classification of osteogenesis imperfecta was by Looser, in 1906 who divided the condition into two forms, osteogenesis imperfecta congenita (also known as Vrolik disease) and osteogenesis imperfecta tarda (also known as Ekman-Lobstein disease).
Since then newer classification systems were based on work by Sillence and colleagues in 1979, have taken into account the phenotypic features and the mode of inheritance. This has since been revised and up to eight forms of osteogenesis imperfecta have been identified.
Three main types are easily distinguished
mild: type I
perinatal lethal: type II
progressive deforming: type III
Types IV to VIII are variable in severity and uncommon (see osteogenesis imperfecta classification for further detail).
Radiographic features
Plain radiograph
This is the preferred initial examination.
-
head, neck and spine
-
chest
accordion ribs
-
pelvis
-
general
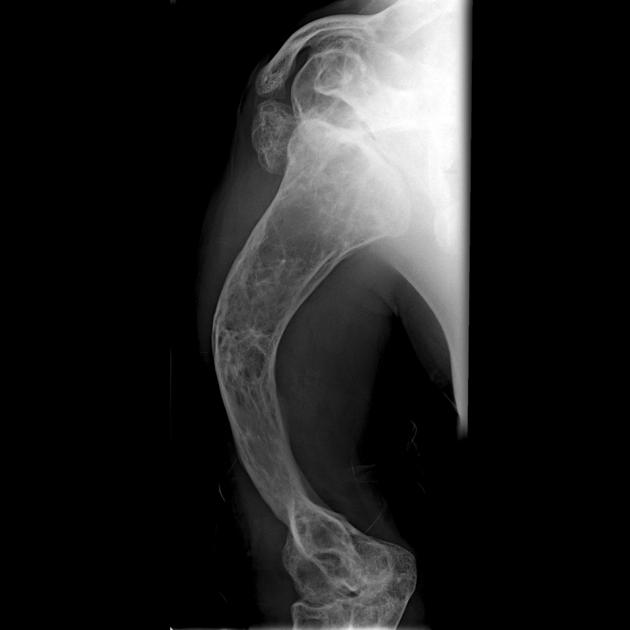
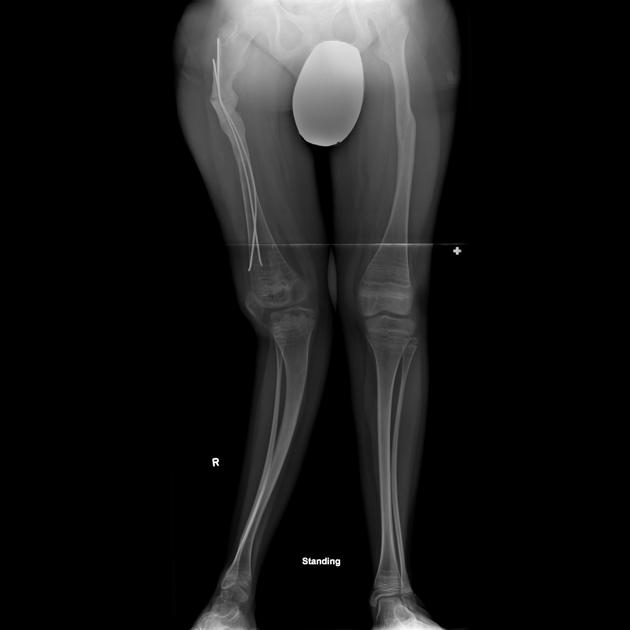
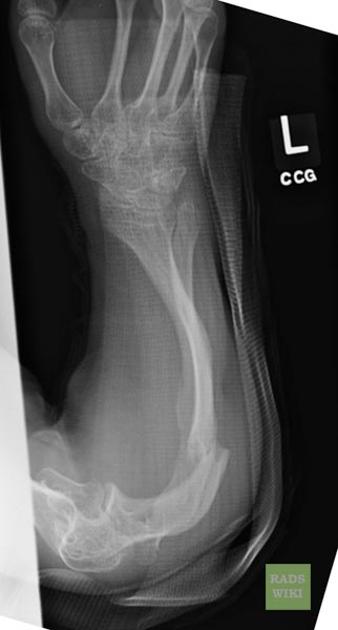
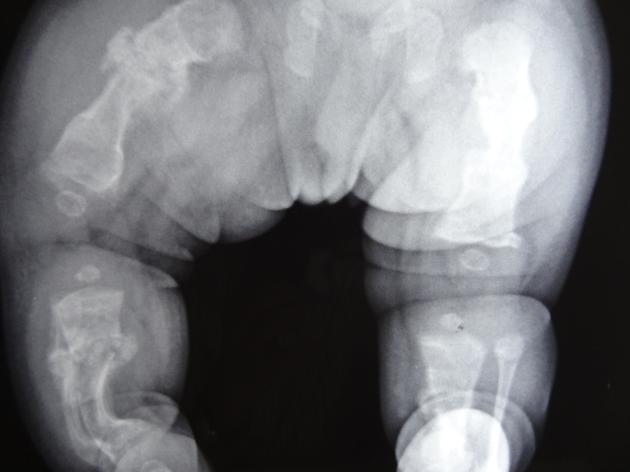
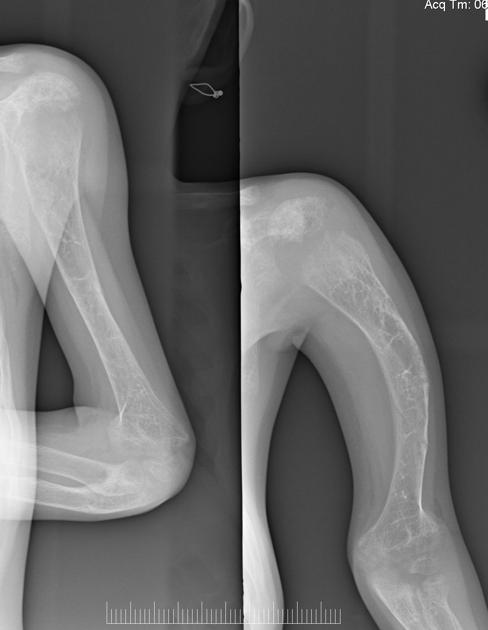
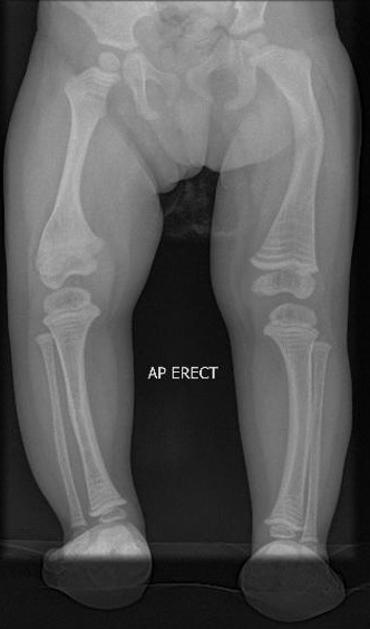
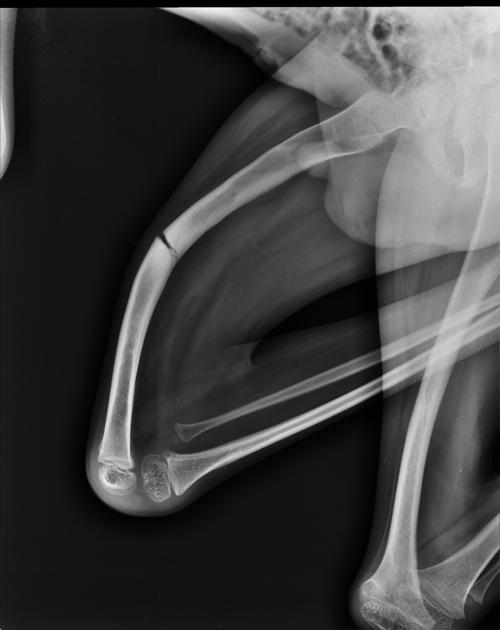
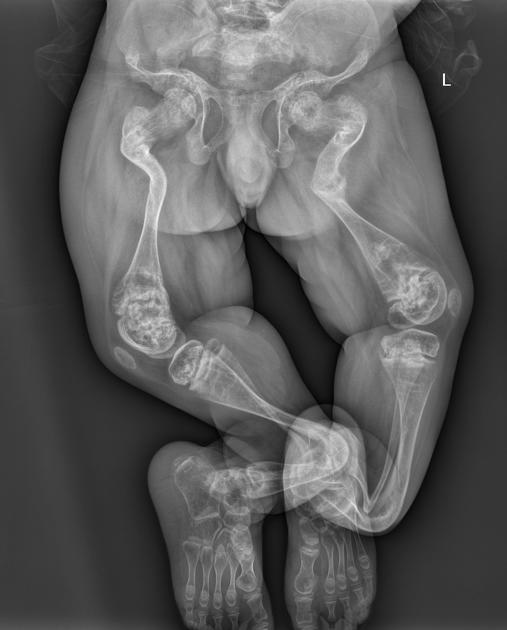
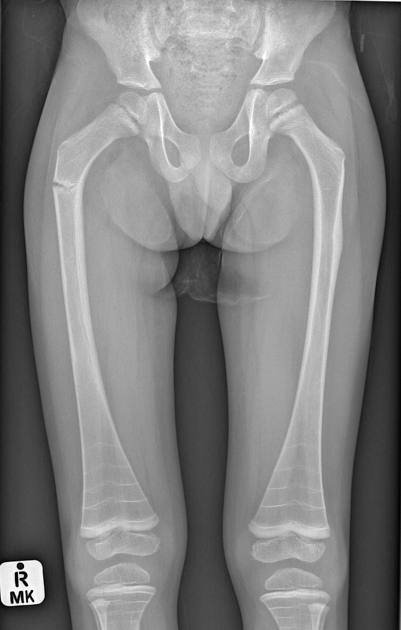
deformed, gracile (overtubulated) bones
cortical thinning
hyperplastic callus formation
popcorn calcification: the metaphyses and epiphyses exhibit numerous scalloped radiolucent areas with sclerotic margins 1
zebra stripe sign: cyclic bisphosphonate treatment produces sclerotic growth recovery lines in the long bones
formation of pseudarthrosis at sites of healing fractures
Prenatal ultrasound
The prenatal sonographic features are often useful in type II (perinatal) and type III forms.
-
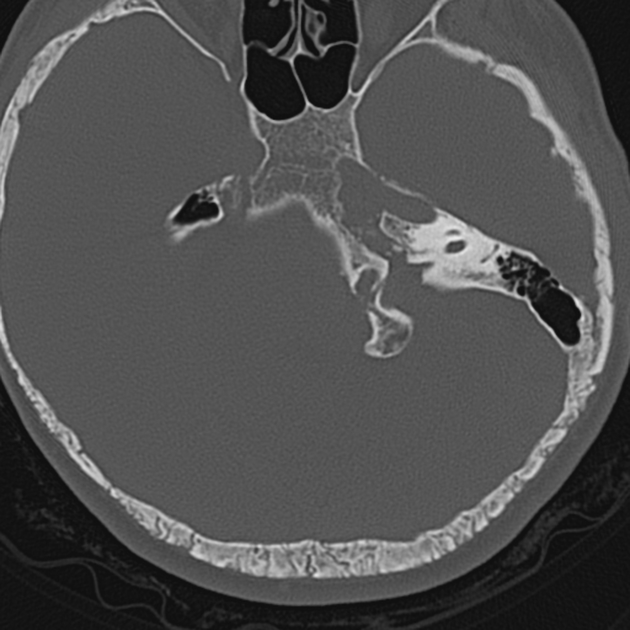
may show hypomineralization of the fetal calvarium
decreased sound wave attenuation and abnormally increased visualization of fetal brain detail
the skull may deform/compress with transducer pressure
-
may show evidence of fractures
long bones may appear shortened and/or angulated as a result
there may be a sonographic gap along the length of a long bone
ribs may have a beaded appearance
there may be the presence of polyhydramnios 8
CT
While showing most of the plain film features, CT may also better demonstrate:
MRI
can be used to assess the extent of basilar invagination
Treatment and prognosis
Prognosis is very variable depending on type ranging from being uniformly lethal from type II to a slight reduction in life expectancy for type I.
Management options for non-lethal types include:
-
surgical correction of deformities and the prevention of fractures
intramedullary rods with osteotomy are used to correct severe bowing of the long bones
intramedullary rods are also recommended for children who repeatedly fracture long bones
different types of rods (surgical nails) are available to address issues related to surgery, bone size, and the prospect for growth; the two major categories of rods are telescopic and non-telescopic 2
bisphosphonates
growth hormone therapy
Differential diagnosis
General considerations on pediatric plain films include:
suspected physical abuse (previously termed non-accidental injury or NAI)
osteopenia of prematurity
Menkes syndrome (or kinky-hair syndrome)