
Tuberous sclerosis (TS), also known as tuberous sclerosis complex (TSC) or Bourneville disease, is a phakomatosis (neurocutaneous disorder) characterised by the development of multiple benign tumours of the embryonic ectoderm (e.g. skin, eyes, and central nervous system).
On this page:
Epidemiology
Tuberous sclerosis has an incidence of 1:6000-12,000, with most being sporadic (see below) 1. Sporadic lymphangioleiomyomatosis (sLAM) is considered to be a forme fruste of tuberous sclerosis 21.
Clinical presentation
Tuberous sclerosis was classically described as presenting in childhood with a pathognomonic triad (Vogt triad) of:
seizures: absent in one-quarter of individuals
intellectual disability: up to half have normal intelligence
adenoma sebaceum: only present in about three-quarters of patients 1
The full triad is only seen in a minority of patients (~30%). Therefore, diagnostic criteria have been developed to aid the diagnosis of tuberous sclerosis.
See tuberous sclerosis diagnostic criteria 2.
When patients do not meet these criteria, they are sometimes referred to as manifesting a forme fruste of the condition.
Pathology
Spontaneous mutations account for 50-86% of cases 3, with the remainder inherited as an autosomal dominant condition. In the majority of such cases (80%) the mutation has been narrowed down to two tumour suppressor genes, both part of the mTOR pathway 3,13:
TSC1: encoding hamartin, on chromosome 9q32-34
TSC2: encoding tuberin, on chromosome 16p13.3 (accounts for most cases)
Radiographic features
Tuberous sclerosis has a significant number of manifestations, involving many organ systems. The most common radiographic manifestations are:
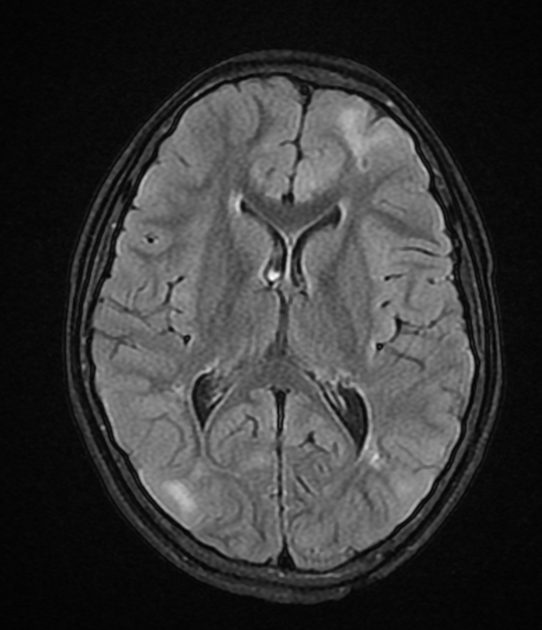
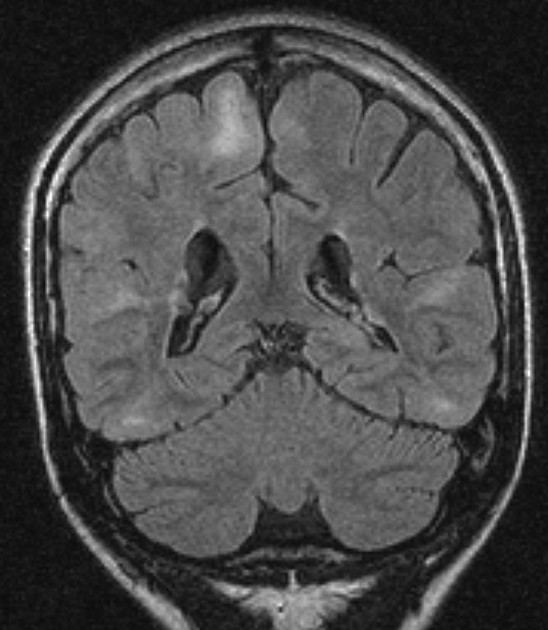
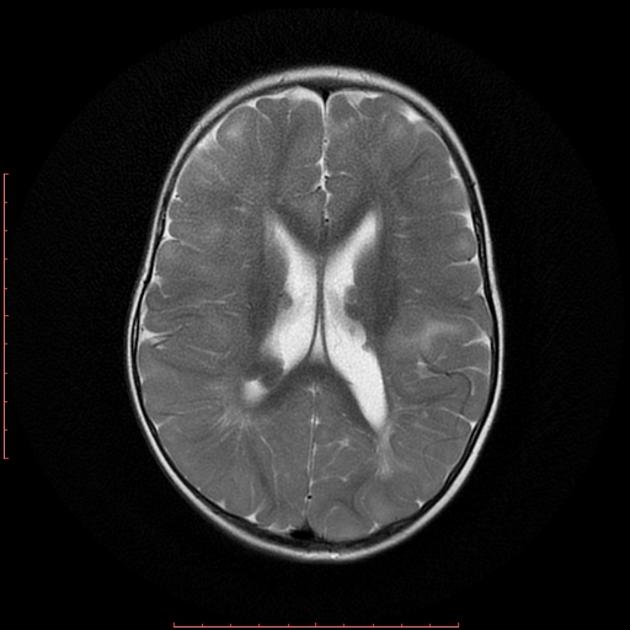
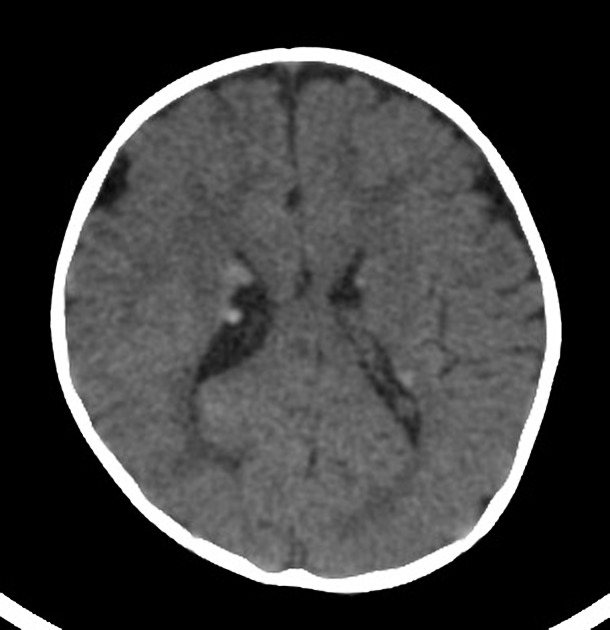
cortical or subependymal tubers and white matter abnormalities
A mnemonic to remember these manifestations is HAMARTOMAS.
Neurological
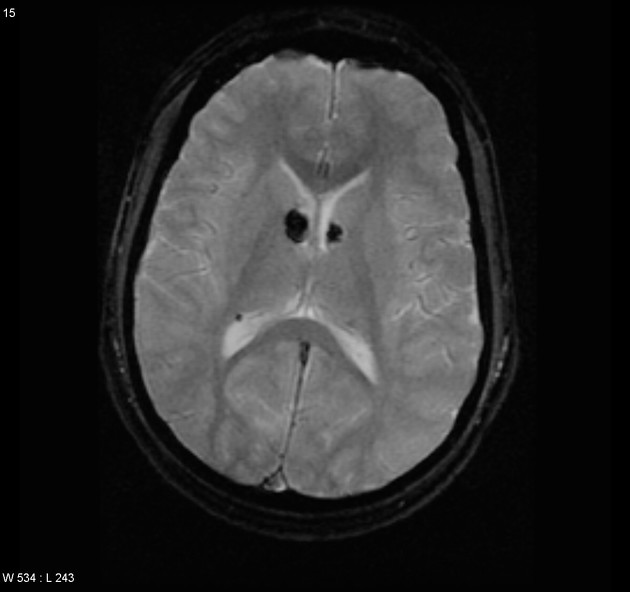
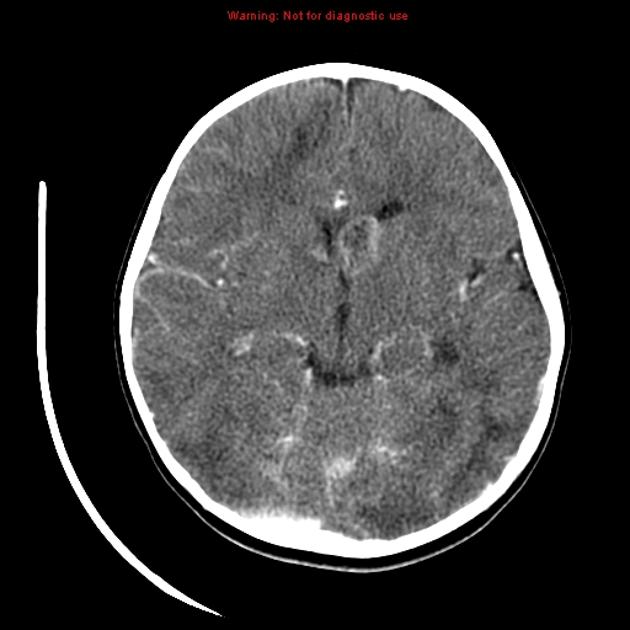
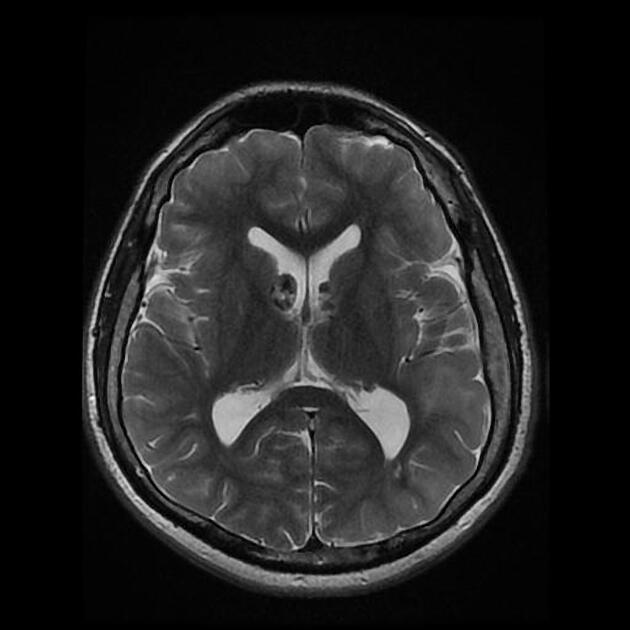
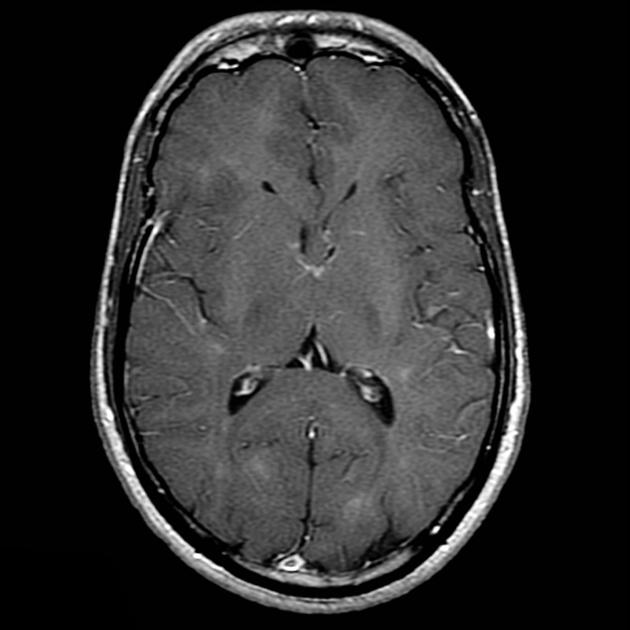
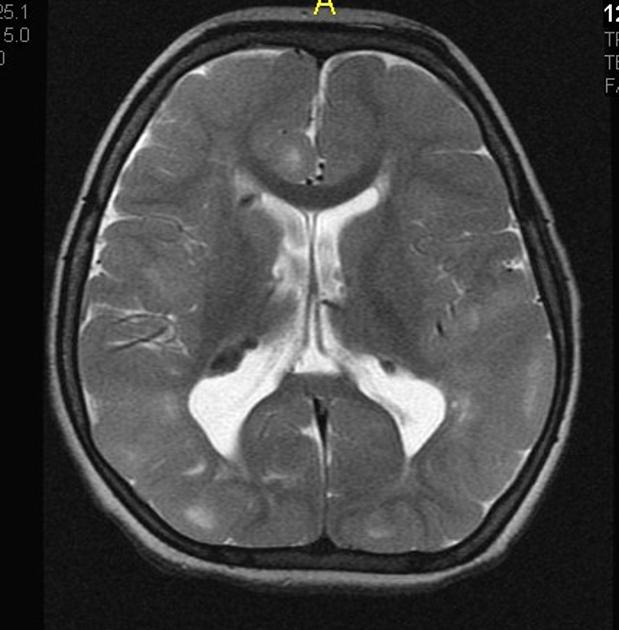
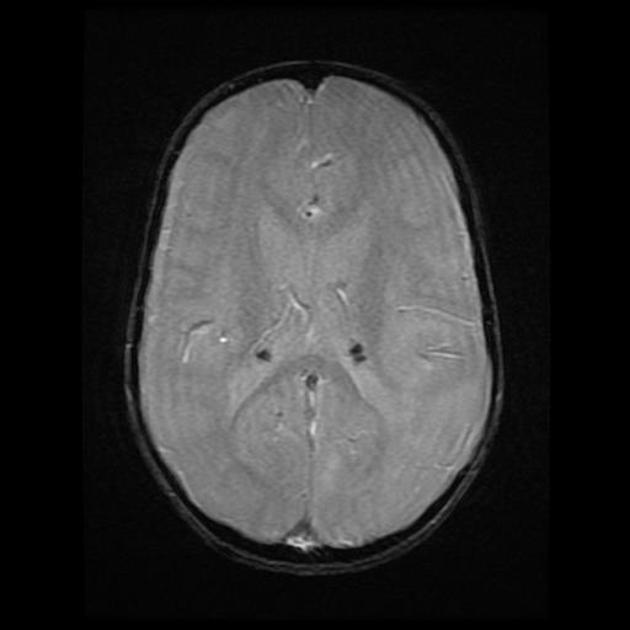
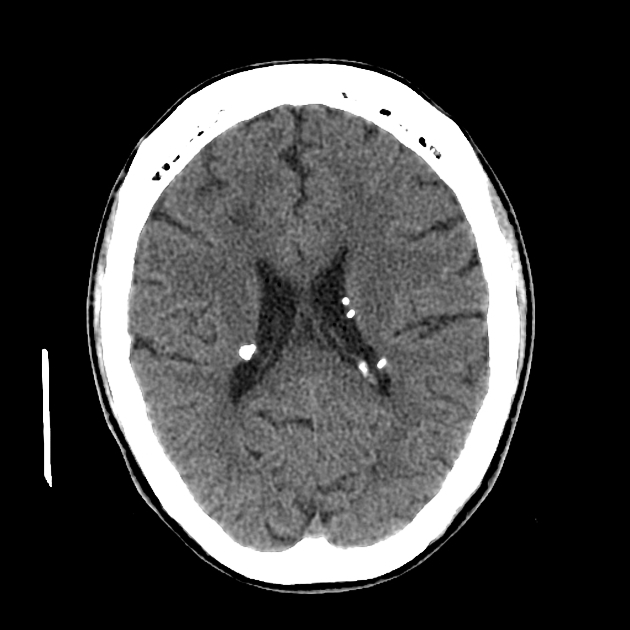
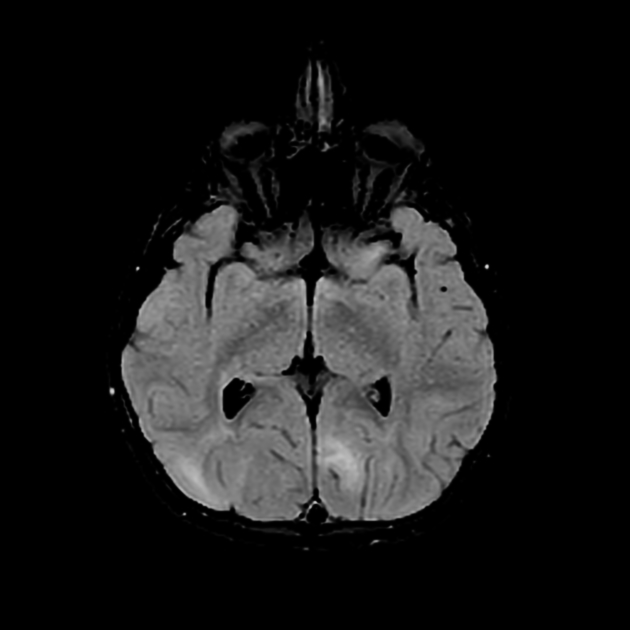
cortical/subcortical tubers: 50% are in the frontal lobe; high T2 and low T1 with only 10% of tubers showing enhancement; frequently calcify after two years of age
-
88% are associated with calcification, although calcification absent in early childhood
visible within the first six months of age 4
variable signal, frequently high T1 and iso to high T2
enhancement is variable and is not a useful feature in distinguishing them from subependymal giant cell astrocytomas (SGCA); only serial growth is reliable 5,6
-
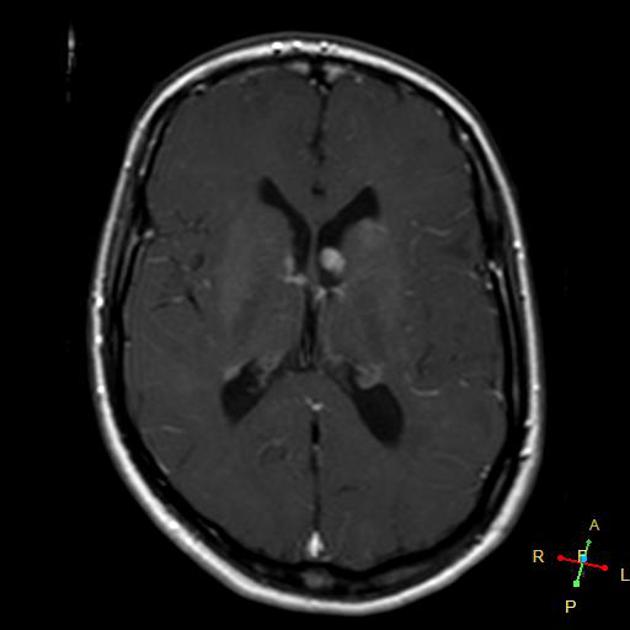
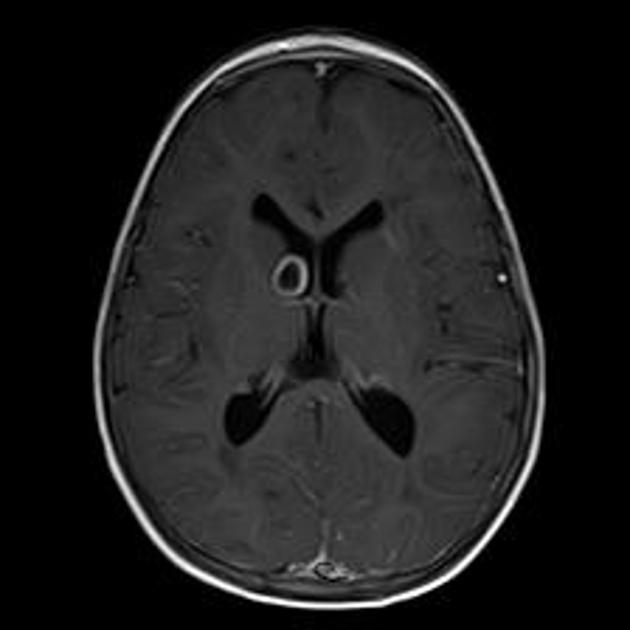
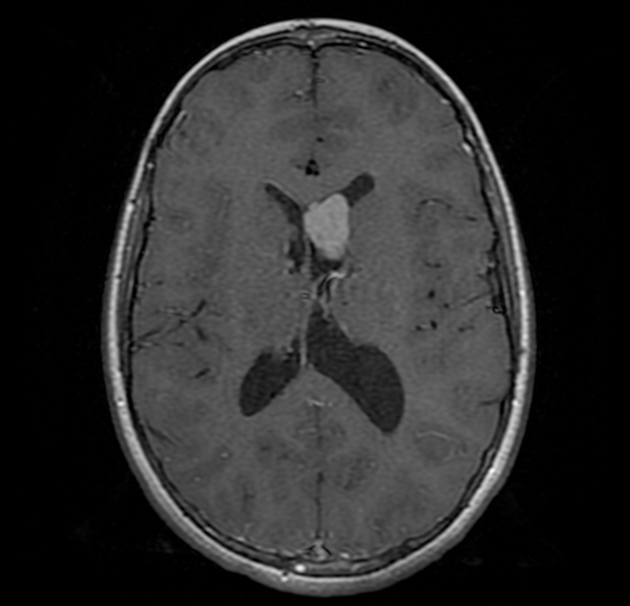
subependymal giant cell astrocytomas (SGCA)
peak occurrence 8-18 years
tend to be large and demonstrate growth 5,6
tend to have intense enhancement
-
white matter abnormalities
variable appearance, with nodular, ill-defined, cystic and band-like lesions seen
radial bands are thought to be relatively specific for TS 7
-
rarer findings
infarcts (due to occlusive vascular disorders)
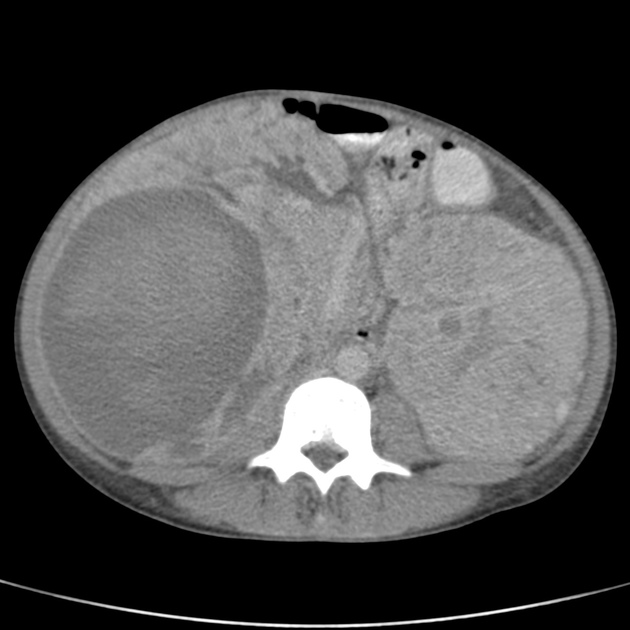
Abdominal
-
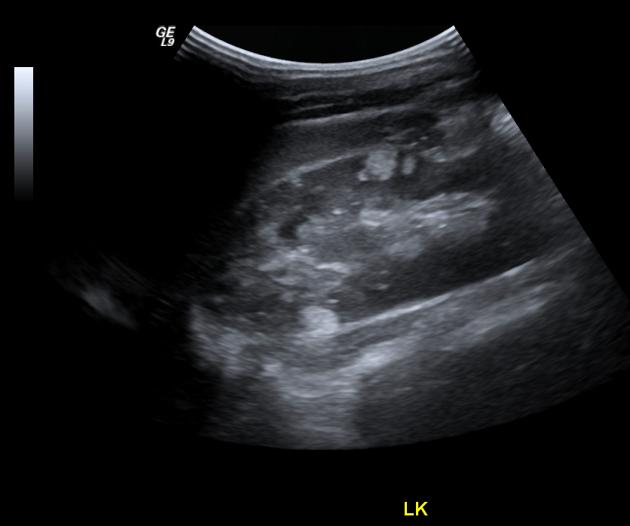
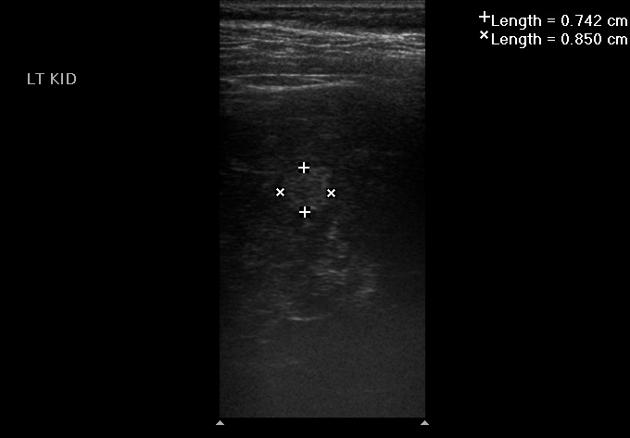
tuberous sclerosis accounts for 20% of all AMLs 3
AMLs are seen in ~65% (range 55-75%) of patients with tuberous sclerosis ref
tend to be multiple, large and bilateral ref
tend to grow and require surgical treatment, as the probability of haemorrhage is proportional to the size ref
micro and macro aneurysms may be present 3
fat may not be visible in up to 4.5% 1
-
renal cysts: the TSC2 gene is located adjacent to the PCKD1 gene 3
18-53% of patients with tuberous sclerosis 1
-
renal cell carcinoma and oncocytomas
although rates of renal cell carcinoma are the same as in the general population, in patients with tuberous sclerosis, renal cell carcinoma tends to occur at a younger age 1
-
retroperitoneal lymphangiomyomatosis (LAM)
histologically identical to pulmonary LAM ref
retroperitoneal cystic lesions ref
chylous ascites, enlarged lymph nodes, dilatation of the thoracic duct ref
gastrointestinal polyps
pancreatic neuroendocrine tumours 12
Thoracic
-
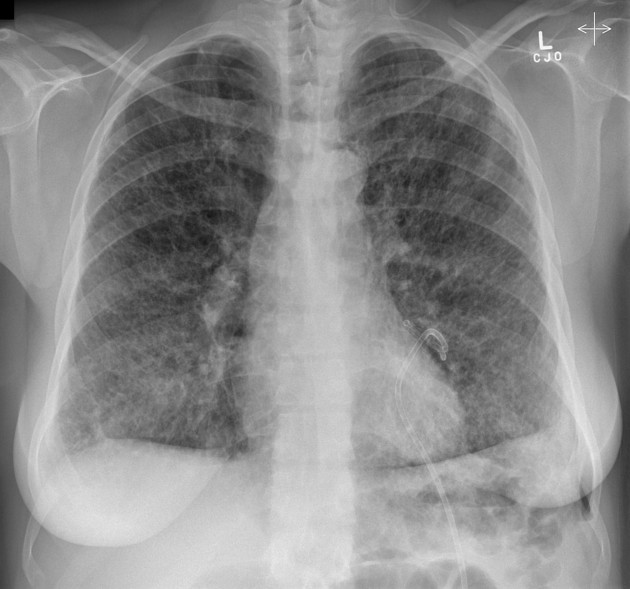
lymphangioleiomyomatosis (LAM)
some studies have described a lymphangiomyomatosis-like change to be present in 25-40% of female patients with tuberous sclerosis
indistinguishable from sporadic LAM, although generally less severe
pneumothorax and chylous pleural effusions common
~80% 10-year survival
-
multifocal micronodular pneumocyte hyperplasia (MMPH)
rare
characterised by multicentric well-demarcated nodular proliferation of type II pneumocytes
benign, non-progressive
differential diagnoses: miliary pulmonary opacities
-
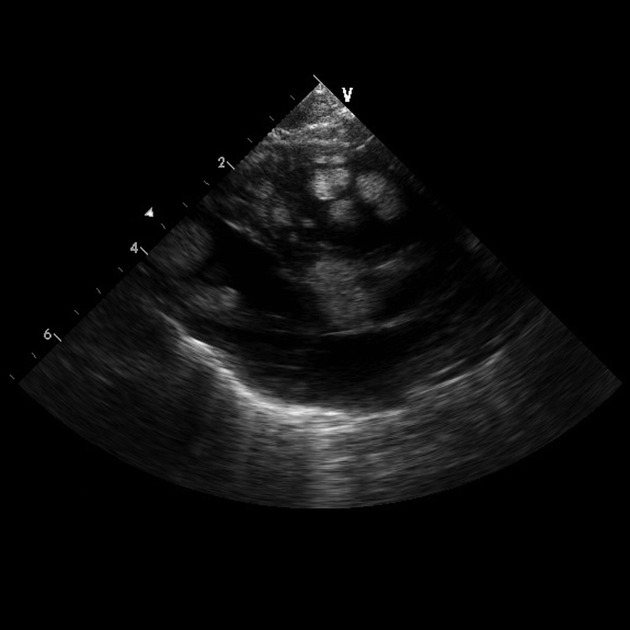
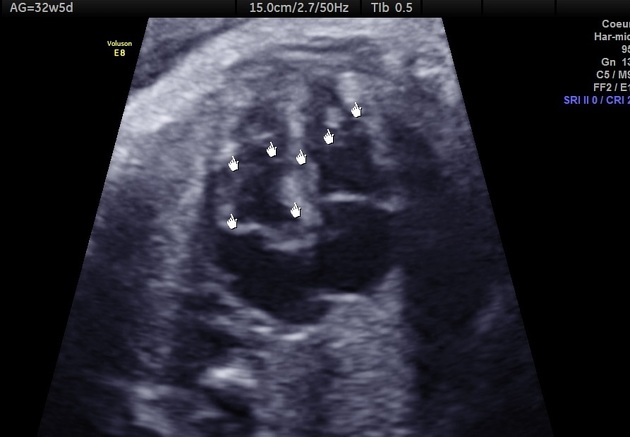
benign cardiac myocyte tumour characterised by the presence of spider cells
seen in 80% of children under 2 years of age with tuberous sclerosis
almost all infants with multiple cardiac rhabdomyomas have tuberous sclerosis, (but only 30% if there is just one tumour)
90% of infants have multiple tumours
multiple ventricular tumours are virtually pathognomonic
depending on size and location, tumours can obstruct inflow or outflow, cause arrhythmias, obstruct a coronary artery or cause fatal congestive heart failure
have been detected as early as 15 weeks gestation and grow through pregnancy, regressing following birth so that the incidence in children over 2 years of age is <20% (tumour growth may be promoted by maternal hormones)
thoracic duct and aortic/pulmonary artery aneurysm
myocardial fatty foci 14 / cardiac fat containing lesions 20
Musculoskeletal
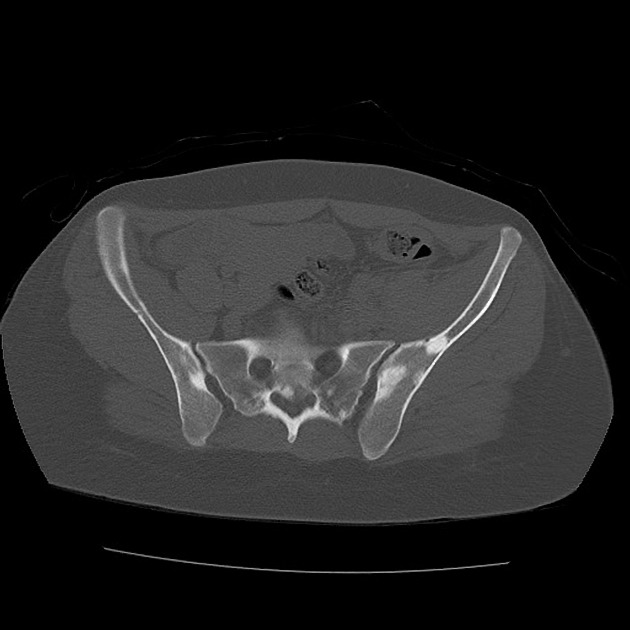
sclerotic bone lesions: ~55% (range 40-66%) 1
hyperostosis of the inner table of the calvaria
periosteal new bone
bone cysts 8
Skin
Cutaneous lesions are present in ~95% of cases, but are rarely appreciated radiographically 8:
hypopigmented macules (ash leaf spots): seen in 90% of patients 1
-
facial angiofibromas (Pringle nodules or adenoma sebaceum); seen in 75% of patients
these skin lesions are neither adenomas nor sebaceous in origin 19
fibrous plaques of the forehead (15-20%)
confetti lesions: variant of leukoderma spots
shagreen patches: seen in 20-30% of patients
periungual angiofibroma (Koenen tumours): 20% of patients
Treatment and prognosis
Treatment of seizures is essential and depending on the degree of intellectual disability, supportive care may be required. Treatment will be dictated by individual manifestations (e.g. subependymal giant cell astrocytomas, or retroperitoneal haemorrhage from renal angiomyolipoma).
Approximately 40% of patients die by age 35 from complications of one or more of the manifestations mentioned above 1.
History and etymology
Historically, the disease was also called Bourneville disease, or even Bourneville-Pringle disease, although both names are still occasionally seen 15,16.
Désiré-Magloire Bourneville (1840-1909) was a French neurologist that is notable for his initial description of tuberous sclerosis (“Bourneville disease”) in 1880. His own medical training included apprenticeship to the renowned French neurologist, Jean Martin Charcot 16.
John James Pringle (1855-1922) was a Scottish dermatologist that also studied this disease leading some books to refer to it as "Bourneville-Pringle disease” 17.
Heinrich Vogt (1875-1957) was a German neurologist who is notable by establishing that the simultaneous presence of three clinical signs was pathognomonic for tuberous sclerosis; this became known as "Vogt triad” 18.










 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.