
Hirschsprung disease is the most common cause of neonatal colonic obstruction (15-20%). It is commonly characterized by a short segment of colonic aganglionosis affecting term neonates, especially boys.
On this page:
Epidemiology
Hirschsprung disease affects approximately 1:5000-8000 live births. In short-segment disease, there is a significant predilection for males (M:F = ~3.5:1), which reduces with the increasing length of involvement 4,6. Interestingly, it is almost never seen in premature infants.
Associations
Although Hirschsprung is an isolated abnormality in 70% of cases, there are some well-documented associations, including 4,6:
Down syndrome: in ~10% of Hirschsprung cases
-
other non-neurocristopathy syndromes
Clinical presentation
The condition typically presents in term neonates with failure to pass meconium in the first 1-2 days after birth, although later presentation is also common. Overall ~75% of cases present within 6 weeks of birth 4 and over 90% of cases present within the first 5 years of life. A minimal number may present in the adult population 1.
In cases of delayed presentation with anorectal constipation, manometry may be useful in distinguishing short/ultrashort segment Hirschsprung disease from other causes 5.
A definitive diagnosis requires a full-thickness rectal biopsy (2 cm above the dentate line as the region below the dentate line usually is aganglionic).
Pathology
Hirschsprung disease is characterized by aganglionosis (absence of ganglion cells) in the distal colon and rectum. It is thought to either occur from a failure of neuroblasts in neural crest cells to migrate into bowel segments or degeneration of already migrated neuroblasts. It affects cells both in the myenteric and submucosal plexuses 4. Hence, functional obstruction develops due to a spasm in the denervated colon.
It is associated with heterozygous loss-of-function mutations in the receptor tyrosine kinase RET.10
It can be anatomically divided into four types according to the length of the aganglionic segment:
-
short segment disease: ~75% *
rectal and distal sigmoid colonic involvement only
-
long segment: ~15%
typically extends to splenic flexure / transverse colon
-
total colonic aganglionosis: ~8% (range 2-13%)
also known as Zuezler-Wilson syndrome
occasional extension of aganglionosis into the small bowel
-
ultrashort segment disease
3-4 cm of internal anal sphincter only
controversial entity
* see notes on percentages
It is postulated that hypoganglionosis (reduced number of ganglion cells) handles intestinal pseudo-obstruction 4.
Radiographic features
Plain radiograph
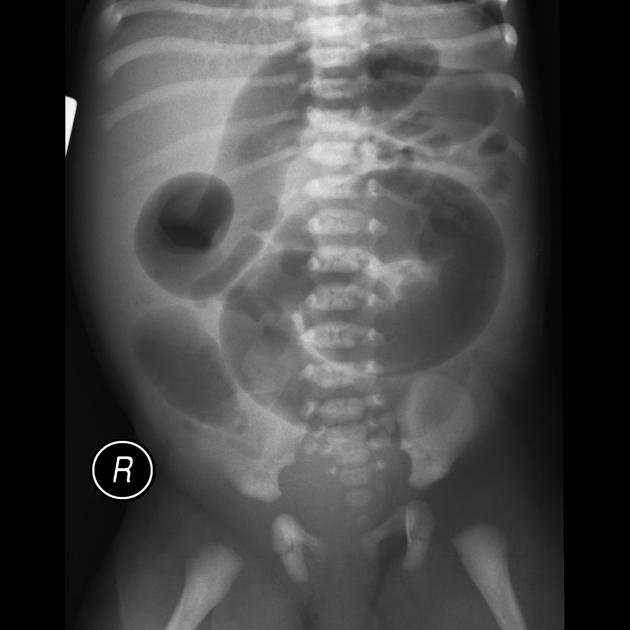
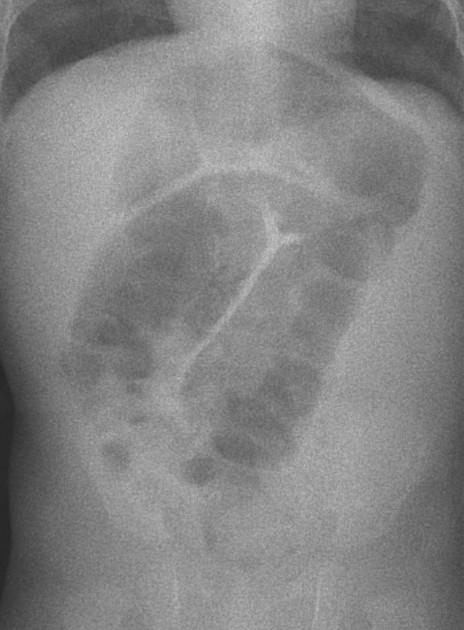
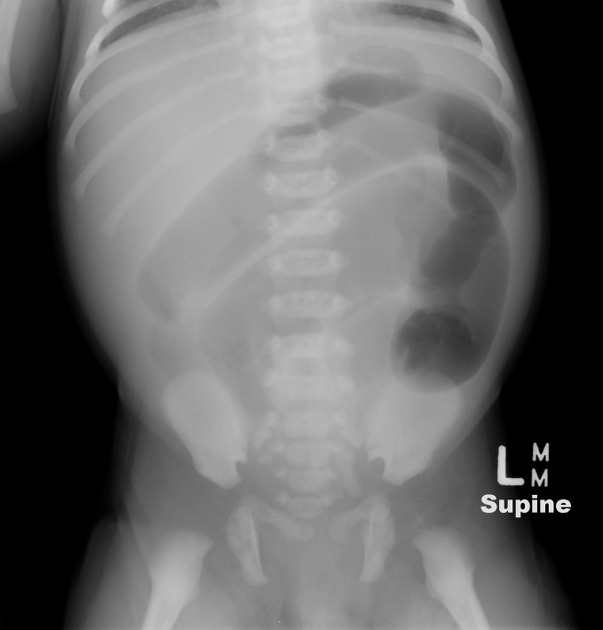
Findings are primarily those of a bowel obstruction. The affected bowel is of smaller caliber; thus, depending on the length of the segment affected, variable amounts of colonic distension are present.
In protracted cases, marked dilatation can develop, which may progress to enterocolitis and perforation.
Fluoroscopy
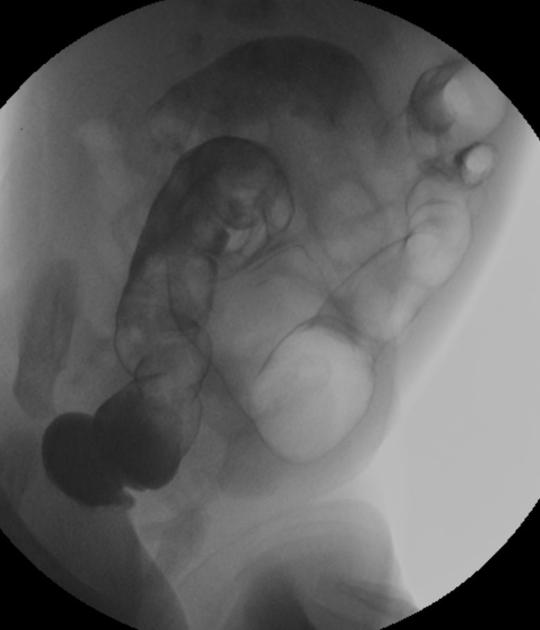
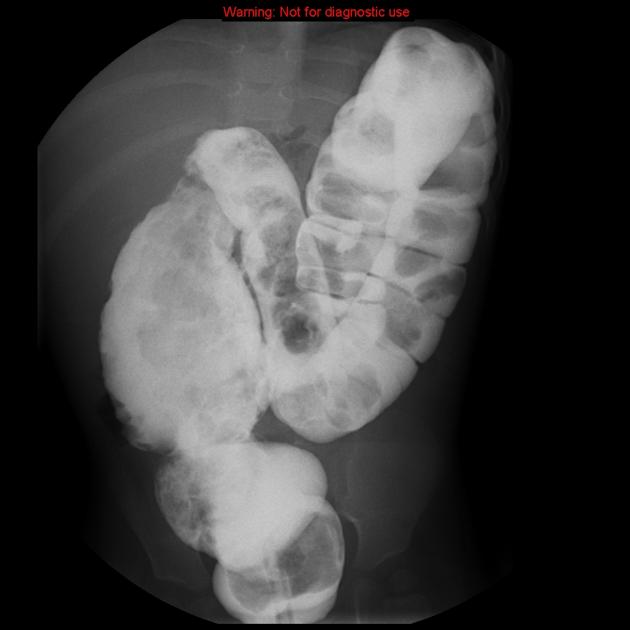
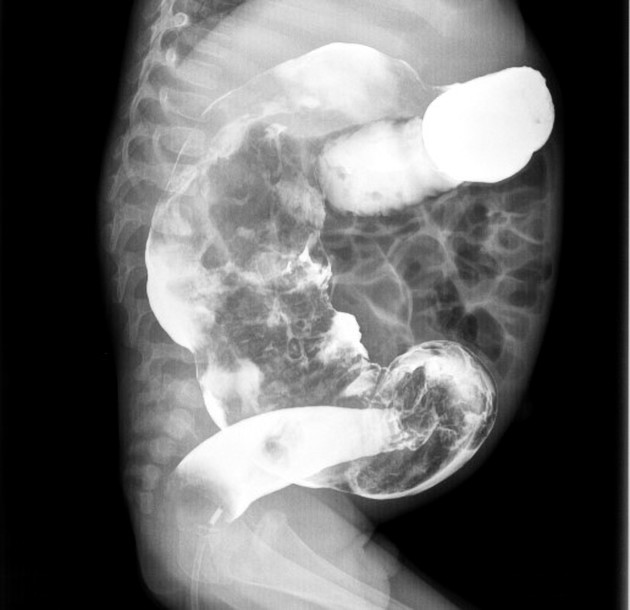
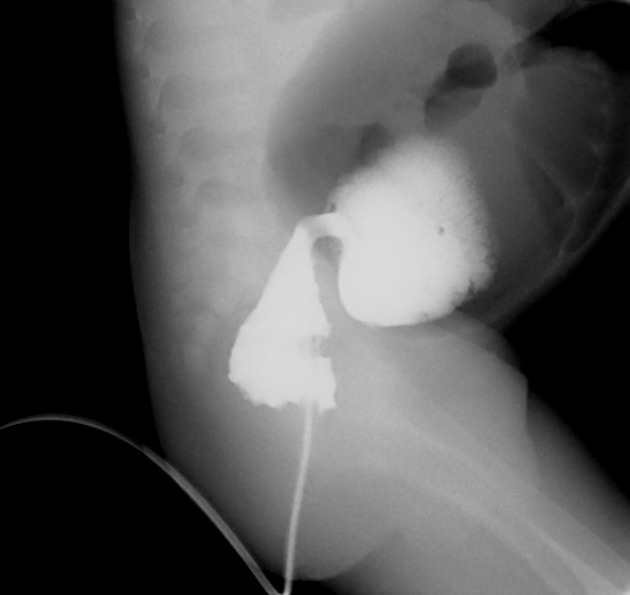
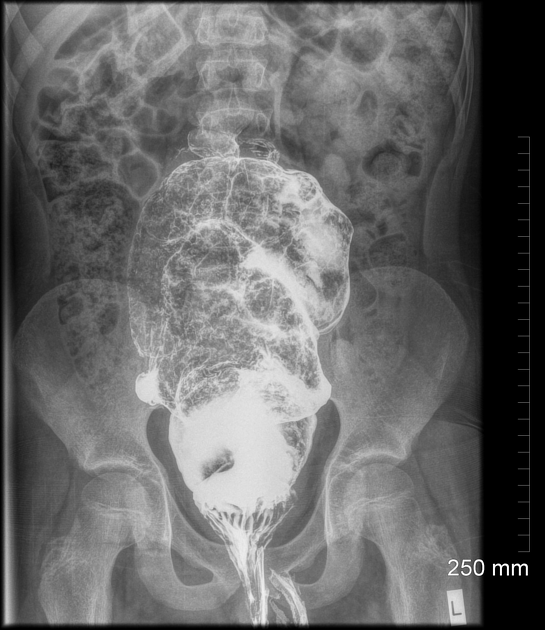
A carefully performed contrast enema is indispensable in both the diagnosis of Hirschsprung disease and in assessing the length of bowel involvement. No bowel preparation is needed before contrast enema. Barium suspension is made with normal saline to avoid fluid absorption by the large surface area of the dilated colon. It should be noted, however, that the depicted transition zone on the contrast enema is not accurate at determining the transition between absent and present ganglion cells.
The affected segment is of small caliber with proximal dilatation. Fasciculation/saw-tooth irregularity of the aganglionic segment is frequently seen. Additionally, delayed evacuation of the administered contrast medium.
Views of particular importance include:
early filling views that include rectum and sigmoid colon, allowing for the rectosigmoid ratio to be determined.
transition zone
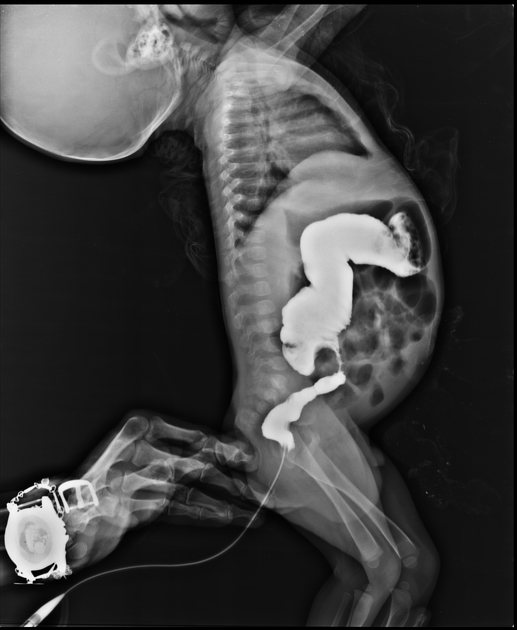
Ultrasound
In particular cases, there may be evidence of fetal colonic dilatation.
Treatment and prognosis
Surgical treatment is by removal of the affected portion of the colon. Where this is successful, the prognosis is good. However, in 3-4% of cases, colonic perforation complicates presentation 2, and this and its sequelae significantly increase mortality and morbidity. Mortality rates can be as high as 30% due to enterocolitis.
History and etymology
It was first described in 1888 by the Danish pediatrician Harald Hirschsprung (1830-1916) 6-8.
Differential diagnosis
General differential considerations include:
microcolon: appears similar to long segment / whole colon Hirschsprung disease







 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}