
Several forms of osteogenesis imperfecta (OI) have been classified, representing a wide variation in appearance and severity, and clinical features vary widely not only between types but within types.
Classification
Osteogenesis imperfecta was initially classified by type according to a scheme developed by David Sillence, an Australian clinical geneticist, based mainly on family history, clinical presentation and radiologic findings. It has since been modified due to the advance in genetics, with the following classification described by Glorieux and Rauch. Further detail can be found on the Osteogenesis Imperfecta Foundation website 1,2.
Type I - mild
Type I is the commonest form (accounting for up to 50% of all cases) and is fortunately also the mildest form. It demonstrates autosomal dominant inheritance. Clinical features include:
general bone fragility and predisposition to fracture
stature is normal or near-normal
loose joints and muscle weakness
bone deformity absent or minimal
-
head and neck features
sclera often have a blue, purple, or grey tint
brittle teeth (dentinogenesis imperfecta) differentiates between the two subtypes IA (without) and IB (with)
predisposed to hearing loss
Type II - perinatal lethal
Type II is the most severe form and is lethal at or shortly after birth mostly due to multiple rib fractures and pulmonary hypoplasia. It results from new dominant mutations to type I collagen genes. Clinical features include:
numerous fractures and severe bone deformity
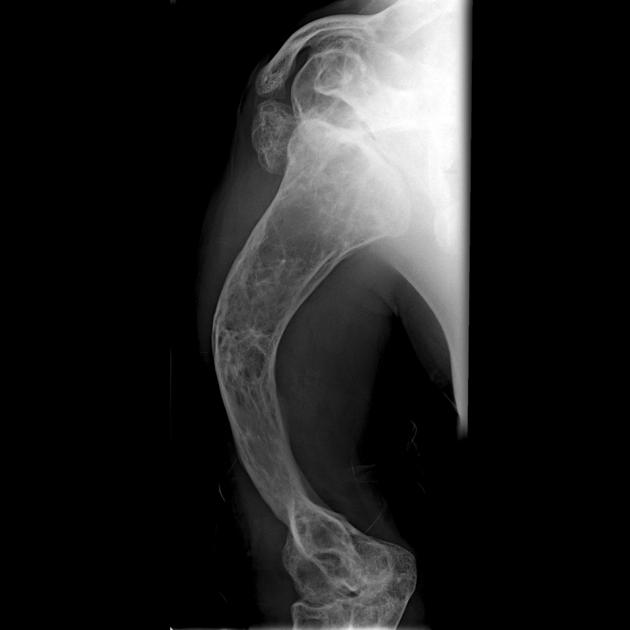
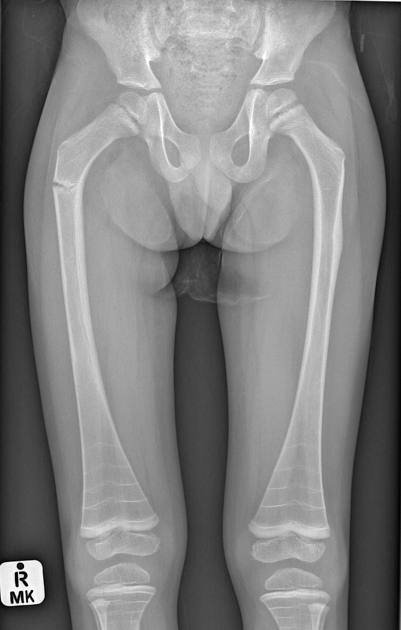
Type III - progressive deforming
Osteogenesis imperfecta type III is the most severe type among children who survive the neonatal period. The degree of bone fragility and the fracture rate vary widely. The majority of osteogenesis imperfecta type III cases result from dominant mutations in type I collagen genes. Clinical features include:
at birth, infants generally have mildly shortened and bowed limbs, small chests, and a soft calvarium
bones fracture easily; fractures are often present at birth, and x-rays may reveal healed fractures that occurred before birth
short stature
rotoscoliosis and vertebral compression fractures.
the altered structure of the growth plates gives a popcorn-like appearance to the metaphyses and epiphyses.
-
head and neck features
sclera have a blue, purple, or grey tint
triangular face
hearing loss
dentinogenesis imperfecta
Type IV - moderate-severe
People with osteogenesis imperfecta type IV are moderately affected. Type IV can range in severity from relatively few fractures, as in osteogenesis imperfecta type I, to a more severe form resembling osteogenesis imperfecta type III. Clinical features include:
bones fracture easily, most before puberty
shorter than average stature for age
mild to moderate bone deformity
-
head and neck features:
sclerae are often light blue in infancy, but the color intensity varies - the sclerae may lighten to white later in childhood or early adulthood
triangular face
dentinogenesis imperfecta possible
hearing loss possible
Type V
Similar to type IV in appearance and symptoms of osteogenesis imperfecta. Dominant inheritance pattern.
Clinical features include:
large hypertrophic calluses at the fracture or surgical procedure sites
calcification of the interosseous membrane between the radius and ulna restricts forearm rotation and may cause dislocation of the radial head
Type VI
Osteogenesis imperfecta type VI is extremely rare. It is moderate in severity and similar in appearance and symptoms to osteogenesis imperfecta type IV and is distinguished by a characteristic mineralization defect seen in biopsied bone. Mode of inheritance is unknown.
Types VII and VIII
Two recessive types of osteogenesis imperfecta, types VII and VIII, have been identified.
Recessively inherited osteogenesis imperfecta has been discovered in people with lethal, severe, and moderate osteogenesis imperfecta. There is no evidence of a recessive form of mild osteogenesis imperfecta. Recessive inheritance probably accounts for <10% of OI cases.
some cases of osteogenesis imperfecta type VII resemble osteogenesis imperfecta type IV in many aspects of appearance and symptoms
other cases resemble osteogenesis imperfecta type II, except that infants have white sclerae, small heads and round faces
cases of osteogenesis imperfecta type VIII are similar to osteogenesis imperfecta types II or III in appearance and symptoms except for white sclerae
Additional forms of osteogenesis imperfecta
The following conditions are rare, but they feature fragile bones plus other significant symptoms 3:
osteoporosis-pseudoglioma syndrome: severe form of osteogenesis imperfecta that also causes blindness
Cole-Carpenter syndrome: osteogenesis imperfecta with craniosynostosis and ocular proptosis
Bruck syndrome: osteogenesis imperfecta with congenital joint contractures
Ehlers-Danlos syndrome: features fragile bones and extreme ligament laxity




 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}