
Pediatric cystic nephromas, previously known as multilocular cystic nephromas, are rare benign renal neoplasms occurring in children. As of the 2016 WHO classification, they are considered distinct from adult cystic nephromas 1,2.
On this page:
Terminology
Evolving terminology regarding cystic nephromas and other cystic renal tumors reflects ongoing changes in classification, in step with shifting understanding of the disease processes and genetic aberrations.
Classically, cystic nephromas were thought to occur in a bimodal age distribution, affecting young children and adults in middle age and the generic term "multilocular cystic renal tumor" was sometimes used.
Lesions arising in children are distinct from adult cystic nephromas on both immunohistochemical and genetic bases, and are separately categorized by WHO 2.
- pediatric cystic nephroma (pediatric)
- cystic nephroma (adult)
- cystic partially differentiated nephroblastoma (pediatric)
The remainder of this article will discuss pediatric cystic nephroma.
Epidemiology
This condition is rare, and occurs in young children (3 months to 5 years of age). There is a 75% male predilection.
Clinical presentation
Pediatric cystic nephroma usually present as an asymptomatic palpable abdominal mass felt by parents, with increasing abdominal girth. It may also present with hematuria or possibly urinary tract infection.
Pathology
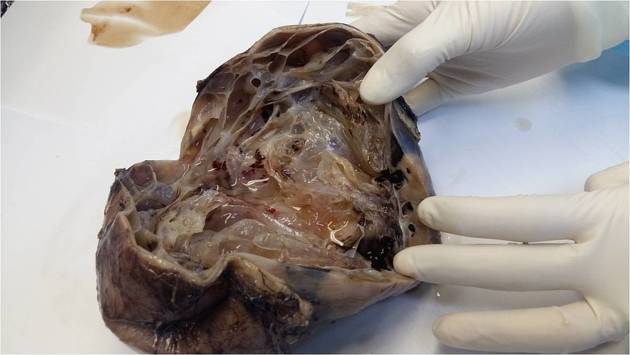
Grossly, cystic nephromas are typically unifocal multiloculated cystic masses surrounded by a thick fibrous capsule and compressed parenchyma 3,4. Calcification, hemorrhage, and necrosis are unusual.
Microscopically, these tumors are lined by flat, cuboid, or hobnail cells 4, while the fibrous septa may contain mature tubules 5. As opposed to cystic partially differentiated nephroblastoma, the tumor septa contain no blastema cells.
Genetic analysis shows a high prevalence (86%) of DICER1 gene mutations in pediatric cases, as compared to ~10% of adult cystic nephroma 2.
Associations
A familial association of cystic nephroma with the cystic type of pleuropulmonary blastoma related to DICER1 mutation has been described.
Radiographic features
Pediatric cystic nephromas generally have the appearance of a multilocular cystic encapsulated mass on most modalities.
Plain radiograph
May present as a large abdominal mass displacing and effacing adjacent bowel loops.
Ultrasound
- multilocular cystic mass originating from kidney 6
- claw or beak-shape of adjacent renal parenchyma (claw sign) may help confirm a renal origin
- cyst contents are usually anechoic, but low-level echoes may be seen
- septal vascularity can also be seen 4
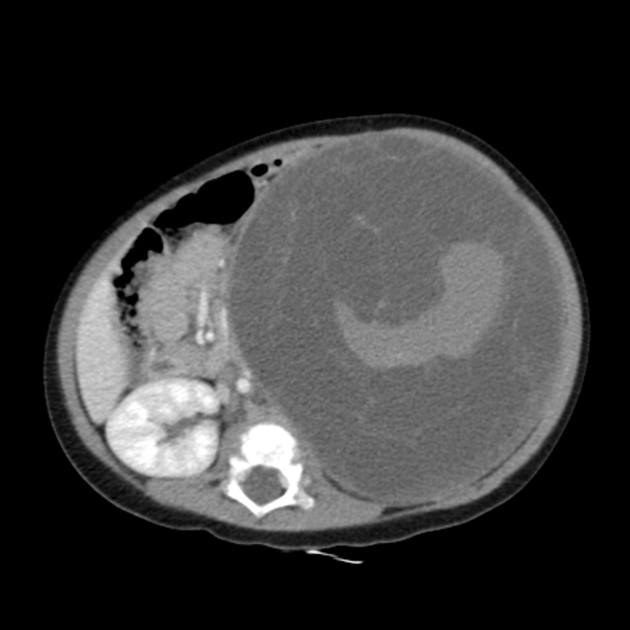
CT
- multilocular cystic mass often herniates into the renal pelvis
- variable septal enhancement
- no nodular or solid enhancement 4
- +/- associated streakiness in perirenal fat 6
MRI
Signal characteristics include 4,6:
- T1: variable signal, depending on the protein or blood products of the cysts
- T2: hyperintense (cysts)
- T1 C+ (Gd): septal enhancement may be seen
Treatment and prognosis
Radical or partial nephrectomy is usually done, with lymph node excision.
Differential diagnosis
-
cystic partially differentiated nephroblastoma
- impossible to differentiate by imaging alone
- similar age group
- no DICER1 mutation
-
cystic Wilms tumor
- solid nodular components in a tumor should arouse suspicion of Wilms tumor 8
- pediatric cystic nephroma tends to occur in younger children than Wilms tumors
-
multicystic dysplastic kidney (MCDK)
- entire kidney is replaced by non-communicating cysts
- usually diagnosed prenatally or at birth, while multiloculated cystic renal tumors do not occur in the perinatal period 4
- enhanced compressed renal parenchyma is seen to surround cystic nephroma on contrast-enhanced cross-sectional studies, while this tissue is absent in MCDK





 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.