
Mayer-Rokitansky-Küster-Hauser syndrome (MRKH), also known as Müllerian aplasia, is a congenital anomaly characterized by vaginal and uterine aplasia or agenesis 9. It is usually also associated with a spectrum of other genitourinary tract abnormalities. MRKH syndrome belongs to class I Mullerian duct anomalies.
Two different forms are described:
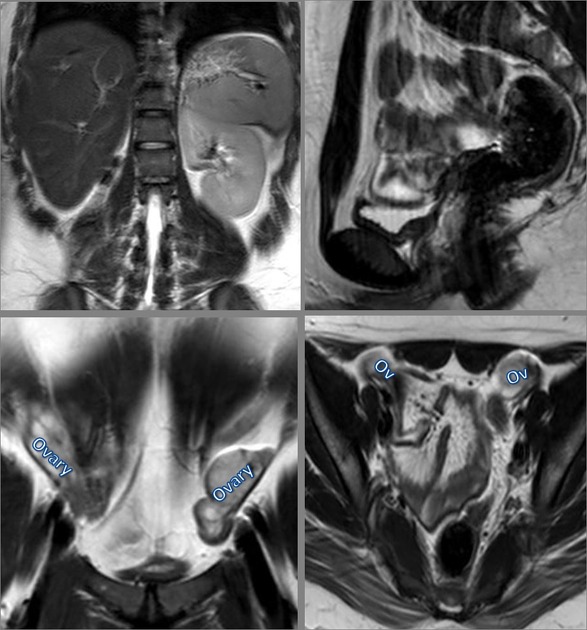
the typical form (type A) of this syndrome is characterized by congenital absence of the uterus and upper 2/3 vagina with normal ovaries and fallopian tubes
the atypical form (type B) of the syndrome includes associated abnormalities of the ovaries and fallopian tubes and renal anomalies
On this page:
Epidemiology
It has a reported incidence of ~1:4000-5000 live female births.
Clinical presentation
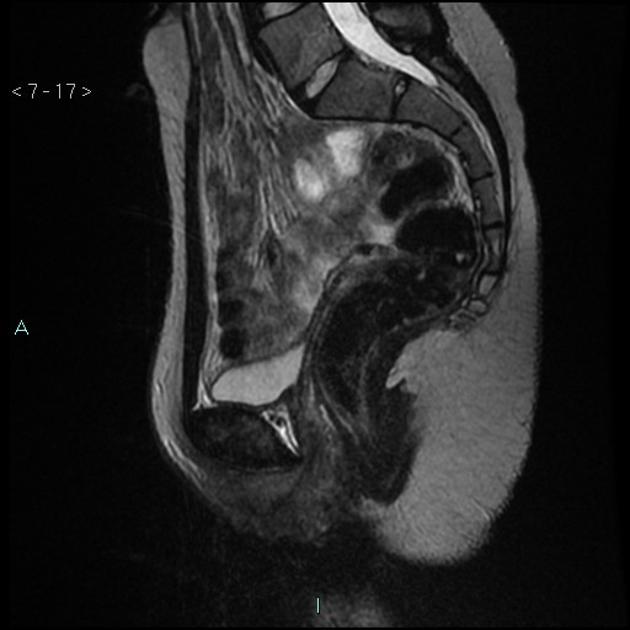
The typical presentation is that of primary amenorrhea, with normal hormonal levels guaranteed by fully functional gonads. At times cyclic pelvic pain may be present in the post-pubertal period due to an accumulation of hemorrhagic material within uterine buds with a functioning endometrium.
Pathology
The anomaly is thought to arise during embryogenesis, with arrested development of the paramesonephric ducts at ~7 weeks after fertilisation.
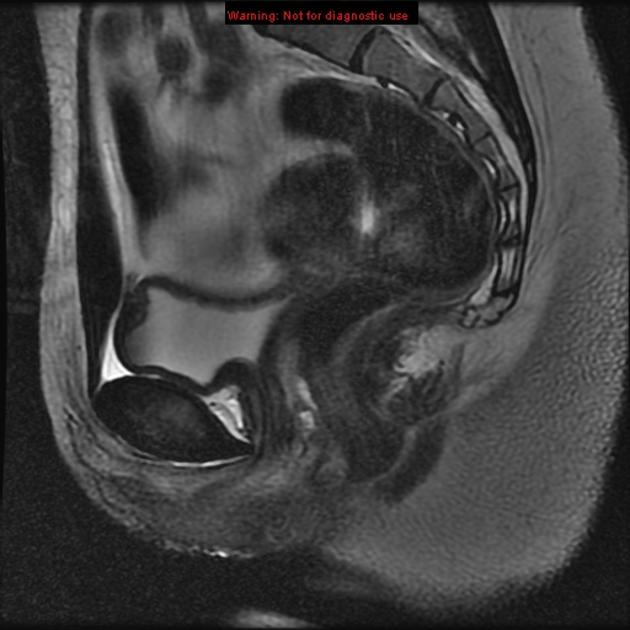
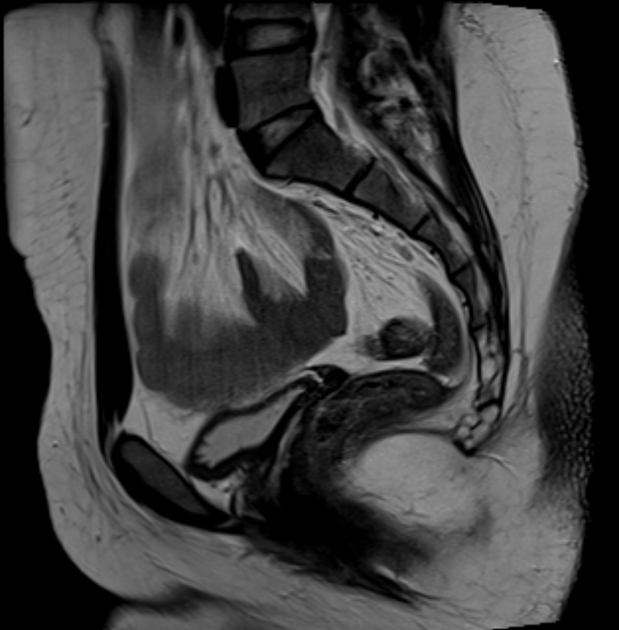
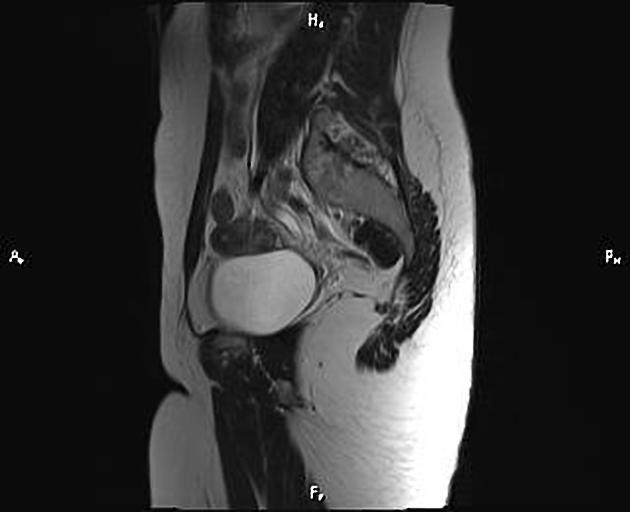
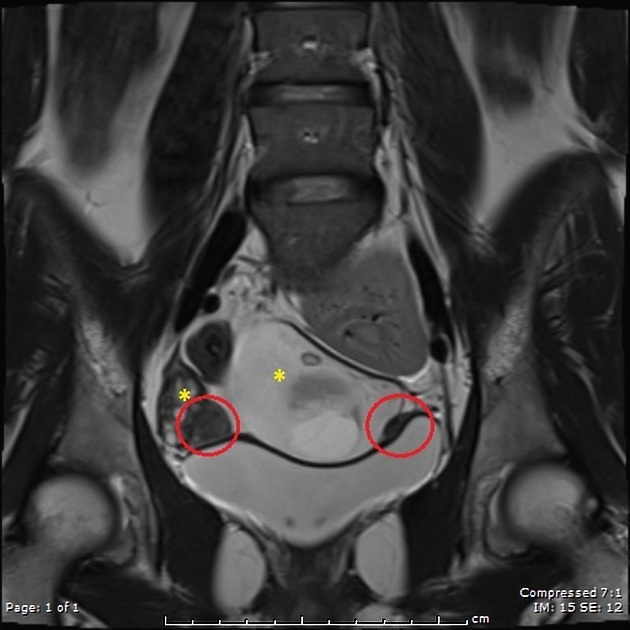
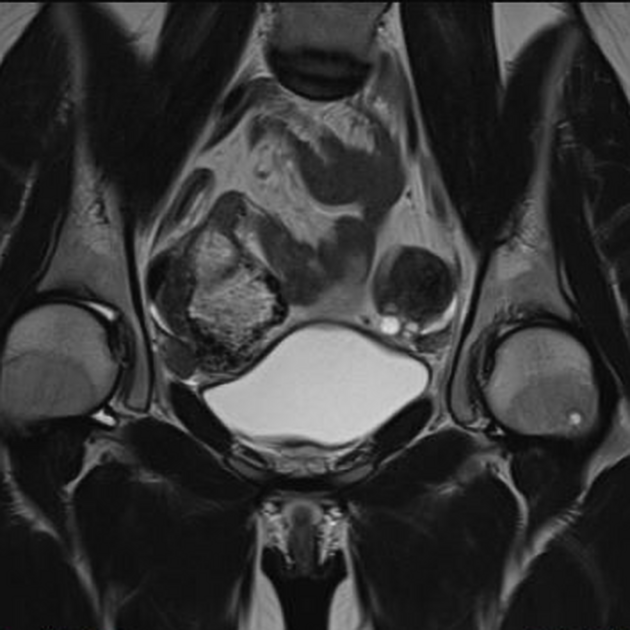
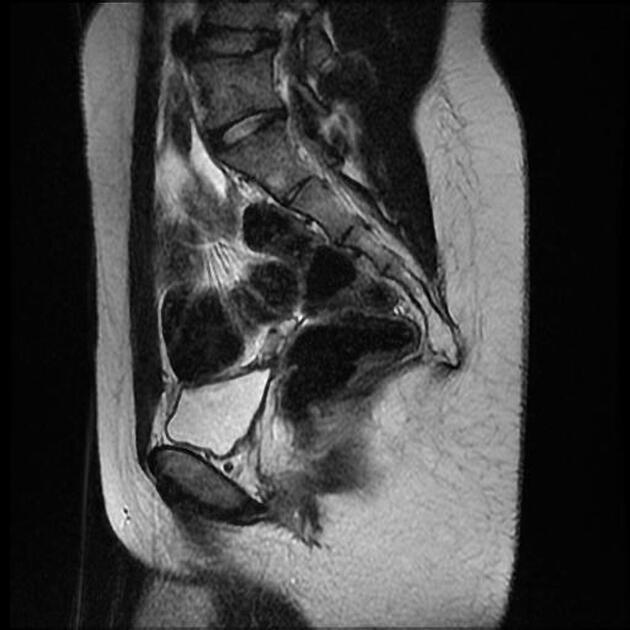
MRKH syndrome is generally characterized by normal external genitalia and absence or reduced development of the uterus and upper two-thirds of the vagina.
The fallopian tubes, uterus, cervix and upper ¾ of vagina develop from the Müllerian ducts from the 8th-12th gestational weeks. A developmental defect occurring at this stage leads to agenesis of Müllerian structures. The development of kidneys, ureter, and bladder occurs concomitantly at around the 6th-12th weeks of gestation. The presence of residual components such as a blind vaginal pouch and a rudimentary uterus (non-functioning myometrial tissue) are noted in a significant proportion of patients.
Associations
The syndrome is often associated with alterations in the urinary or skeletal system which include:
vertebral anomalies: may be present in ~10% of cases
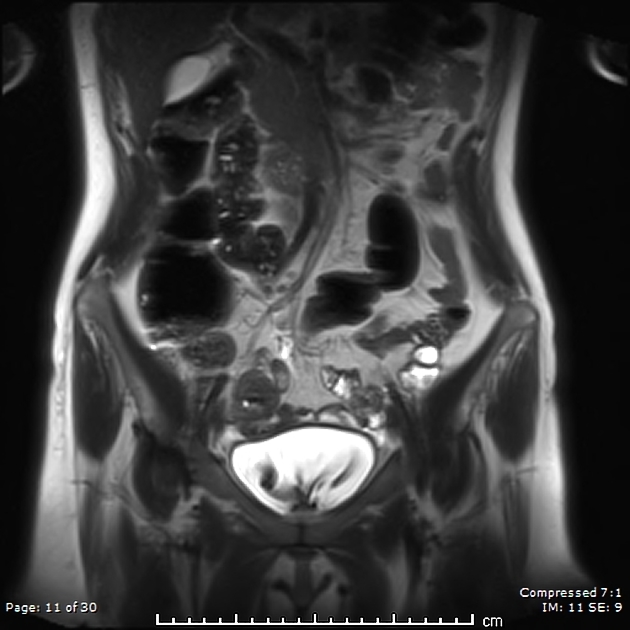
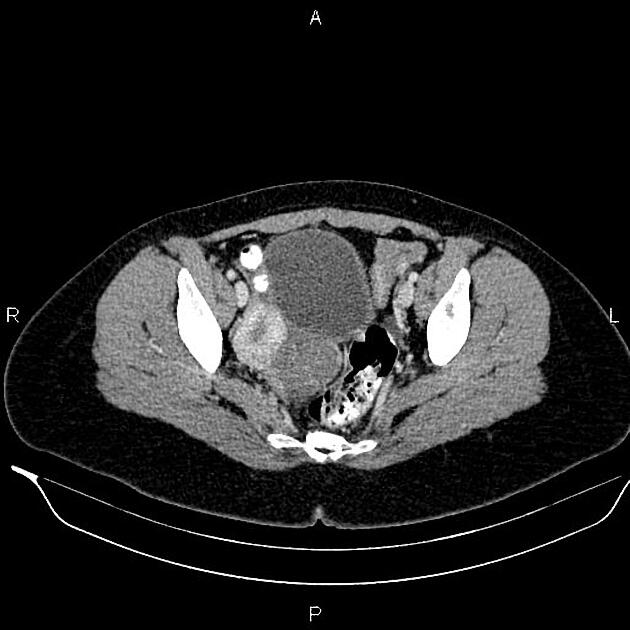
renal anomalies: such as renal agenesis, ectopic kidney, fused kidney, renal hypoplasia, and horseshoe kidney may be seen in 30-40% of patients with the syndrome 4
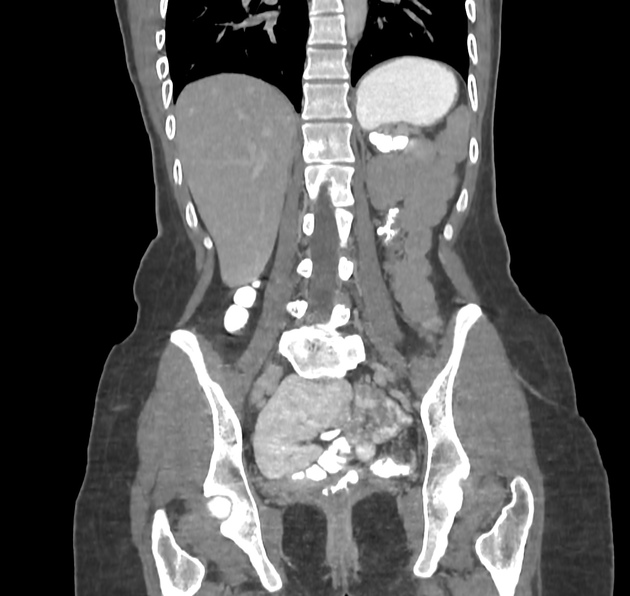
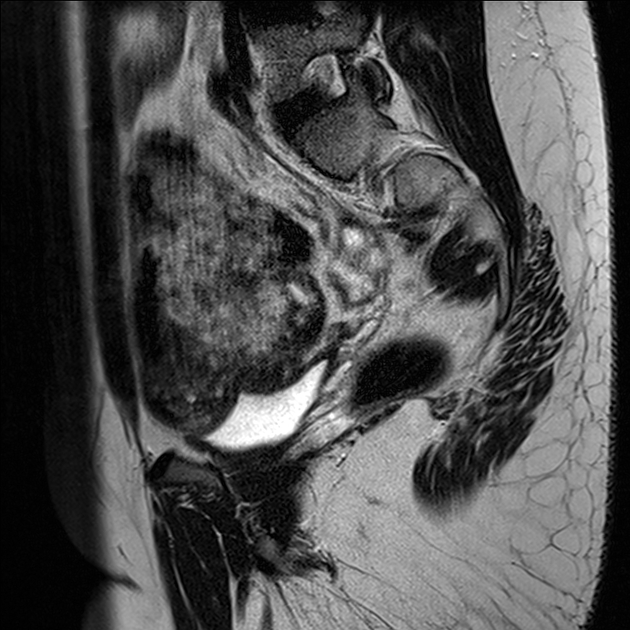
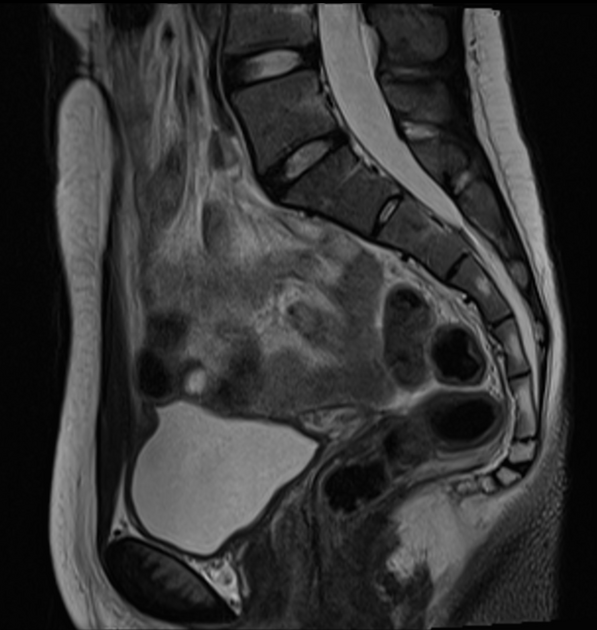
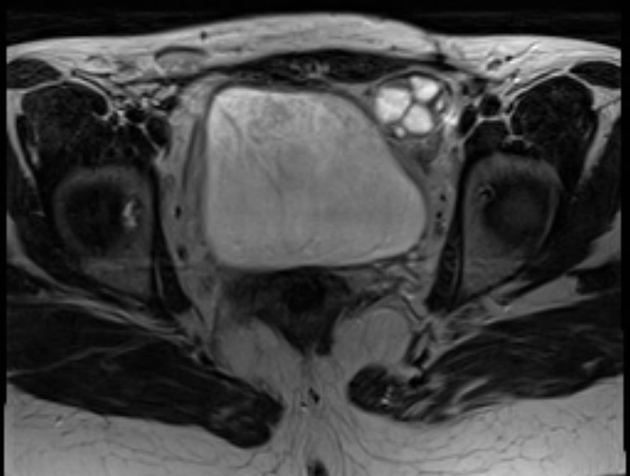
Radiographic features
Hysterosalpingography
Although hysterosalpingography has a well-established use in characterizing uterine Mullerian duct anomalies, it has no place in MRKH syndrome given the hypoplasia/agenesis of the uterus and 2/3 of vagina.
Ultrasound
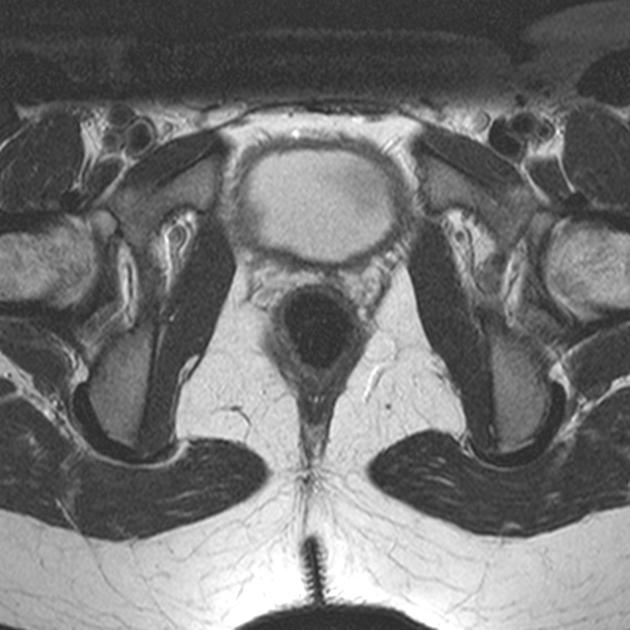
Usually there is absence of the uterus and normal ovaries. Also, ultrasound may demonstrate any associated renal tract anomaly.
MRI
This is the preferred imaging modality following the initial assessment with ultrasound, as it allows for the characterization of the uterine buds and the determination of the presence of functioning endometrium within them, and also any related renal tract anomaly.
History and etymology
It is named after the German physician August Franz Josef Karl Mayer (1787-1865), Austrian pathologist Karl Freiherr von Rokitansky (1804-1878), German gynecologist Hermann Küster (1879-?) and the Swiss physician Georges André Hauser (1921-?) 7,8.





 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.