
Peutz-Jeghers syndrome is one of the polyposis syndromes. It has an autosomal dominant inheritance and is characterized by:
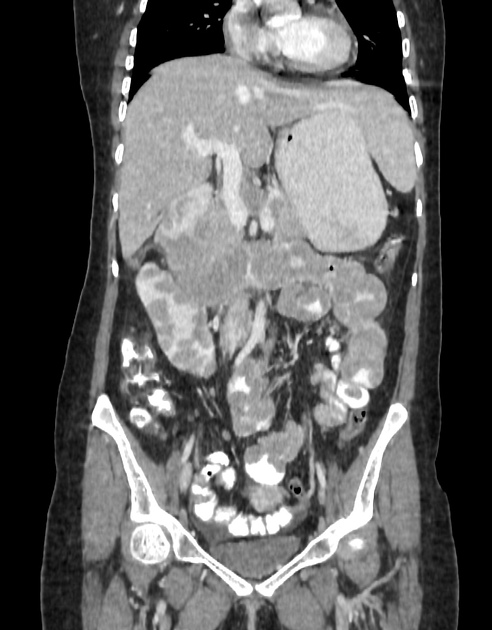
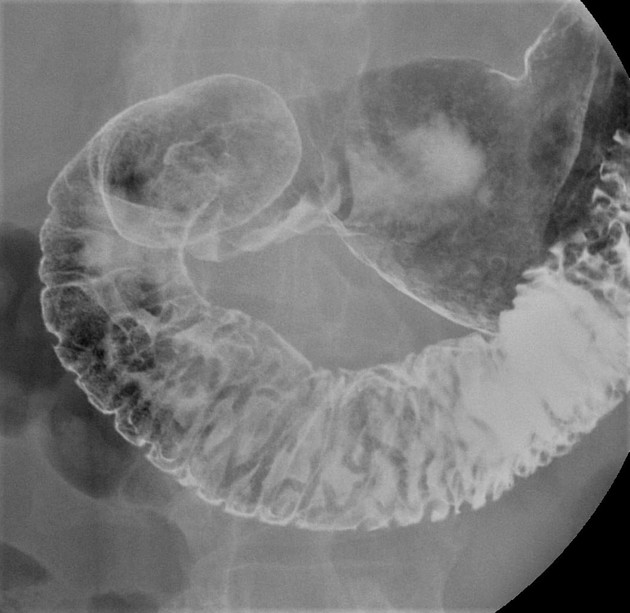
multiple hamartomatous polyps, most commonly involving the small intestine (predominantly the jejunum), but also colon and stomach; mouth and esophagus are spared
mucocutaneous melanin pigmentation involving the mouth, fingers and toes
On this page:
Epidemiology
Peutz-Jeghers syndrome has been reported to be as common as 1 in 8300 live births.
Clinical presentation
Findings on clinical examination include mucocutaneous hyperpigmented macules of the nose, buccal mucosa, axilla, hands, feet and genitalia 4. A clinical diagnosis can be made following histopathological confirmation of typical Peutz-Jeghers syndrome morphology in 2 or more intestinal polyps or after any number of polyps or hyperpigmented macules (in a characteristic location) with a positive family history 4.
Pathology
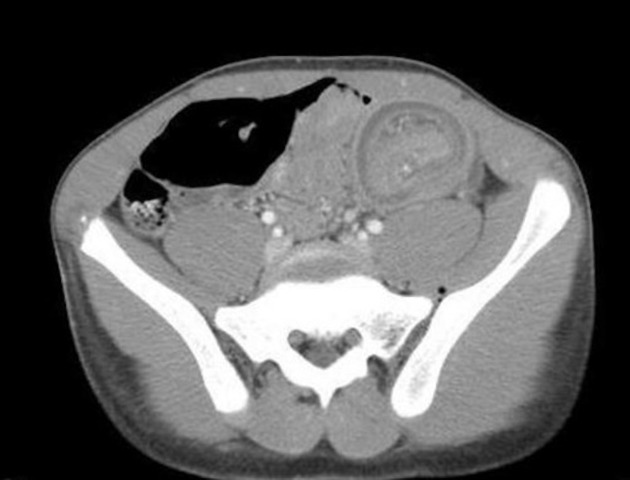
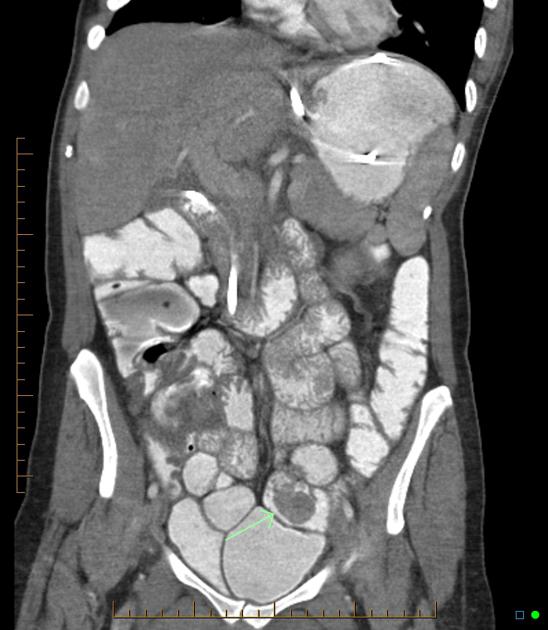
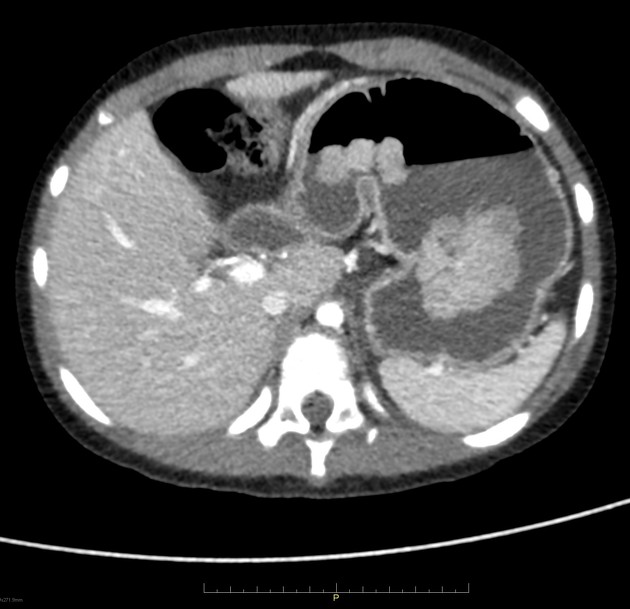
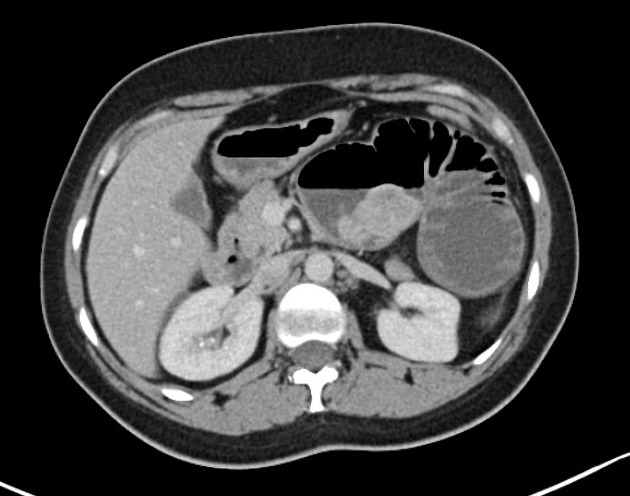
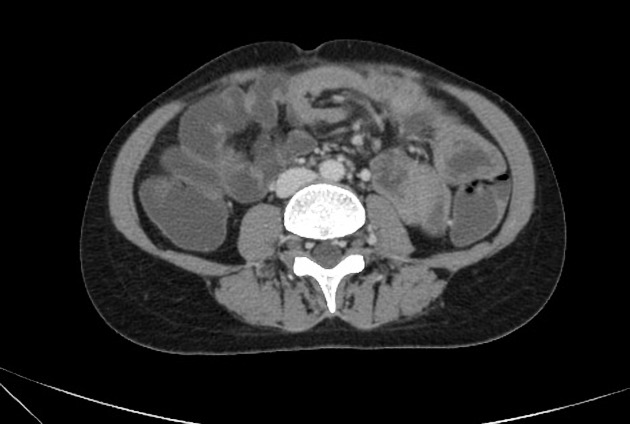
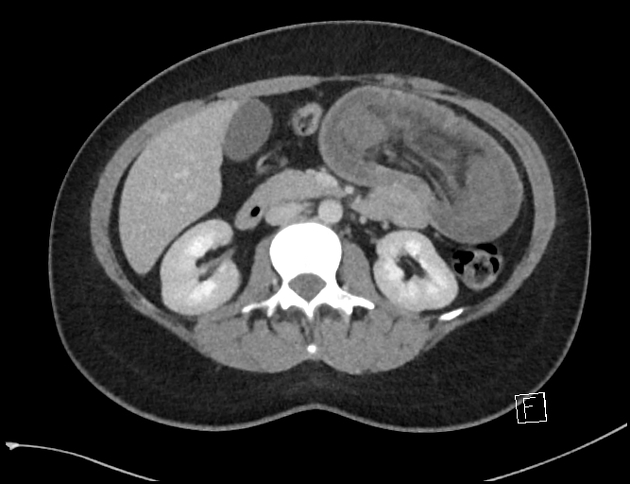
Peutz-Jeghers polyps are non-neoplastic hamartomas due to the proliferation of all three layers of the mucosa, which have a characteristic feature of a smooth muscle core continuous with muscularis mucosa in a tree-like branching pattern. This distinguishes them from the hamartomatous polyps of Cronkhite-Canada syndrome, juvenile polyposis and Cowden disease 1.
Patients are at increased risk of:
-
GI tract adenocarcinoma, although the polyps themselves are not premalignant
colorectal: 39% lifetime risk 5
stomach: 29% lifetime risk 4
small intestine: 13% lifetime risk 4
-
extraintestinal malignancies
adenoma malignum (adenocarcinoma subtype of the cervix)
breast: 45-50% lifetime risk 4, more frequently ductal
pancreas: 11-36% lifetime risk 4
ovary: 18-21% lifetime risk 4, mainly sex cord tumors
uterus: 9-10% lifetime risk 4
cervix: 10-23% lifetime risk 4
testis: 9% lifetime risk 4, large calcifying Sertoli cell tumors
lung: 15-17% lifetime risk 4
Genetics
It is attributed to mutations in tumor suppressor genes, most commonly STK11 (70-94%) 4.
Treatment and prognosis
Due to the increased risk of malignancy, screening is generally recommended. Examples include annual mammography and contrast-enhanced breast MRI, beginning at 25 years of age; baseline CT/MR enterography at 8-10 years of age (and every 2-3 years from 18 years of age); MRCP or endoscopic US every 1-2 years beginning from 30-35 years of age 4.
History and etymology
The syndrome is named after Jans Peutz (1886-1957), a Dutch physician and Harold Jeghers (1904-1990), an American physician who had successively described the association between polyposis and the mucocutaneous macules.


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.