
Aicardi-Goutières syndrome is a rare hereditary neurodegenerative disease which usually presents in early infancy as a systemic and central nervous system inflammatory syndrome characterized by hepatosplenomegaly, vasculopathy and encephalopathy. Many of the features are similar to congenital TORCH infections.
On this page:
Terminology
Evidence suggests that familial systemic lupus erythematosus and microcephaly-intracranial calcification syndrome (MICS) (also known as pseudo-TORCH syndrome or Baraitser-Reardon syndrome) are in fact phenotypic variants of Aicardi-Goutières syndrome 1.
Note: Aicardi-Goutières syndrome is distinct from Aicardi syndrome.
Epidemiology
Aicardi-Goutières syndrome is rare and usually inherited in an autosomal recessive pattern. Penetrance is variable as is the age of clinical onset; most occur within the first year of life (3-7 months is typical) although delayed onset in later childhood is reported 2.
Clinical presentation
Aicardi-Goutières syndrome has variable penetrance with some individuals being only mildly affected with normal intelligence. The majority, however, present with both physical and intellectual disabilities in infancy after a period of apparently normal development (often only lasting a few weeks).
Presentation may be with 1,2:
-
neurological features
developmental delay and regression
irritability
ocular and myoclonic jerks
seizures (25%)
small head size
intermittent pyrexia
hepatosplenomegaly
abnormal liver enzymes
thrombocytopenia
chilblain of the fingers/toes and ears
During the active phase of the disease, elevation of interferon-α levels and lymphocytosis can be detected in the cerebrospinal fluid (CSF) 1,2.
As the disease progresses, additional symptoms may include but are not limited to glaucoma, hypothyroidism, pulmonary hypertension, immune hepatitis, myopathy, and arthropathy 8,9.
Pathology
Aicardi-Goutières syndrome is classified as an interferonopathy; these are a group of autoinflammatory diseases characterized by mutations affecting interferon signaling pathways 3.
In Aicardi-Goutières syndrome mutations occur that result in impaired degradation of cellular nucleic acid debris. Accumulation of this debris eventually triggers an interferon-mediated immune response, with ensuing damage to various tissues similar to that seen in antenatal infections (e.g. TORCH) and autoimmune diseases (e.g. systemic lupus erythematosus) 2.
Aicardi-Goutières syndrome can be the result of a number of genetic mutations. These usually, but not invariably, demonstrate autosomal recessive inheritance 1,5.
Multiple genes have been identified including:
autosomal recessive or dominant: ADAR, TREX1 (common)
autosomal recessive: RNASEH2A, RNASEH2B (most common), RNASEH2C, SAMHD1, LSM11, RNU7-1
autosomal dominant: IFIH1
Radiographic features
CT
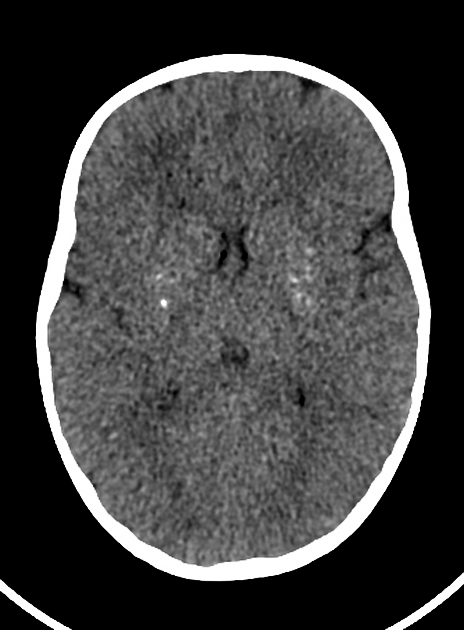
CT shows calcification of the basal ganglia, thalamus and paraventricular white matter 1. The morphology of the calcification varies greatly from large "chunks" of dense calcification to tiny punctate specks 1,2.
MRI
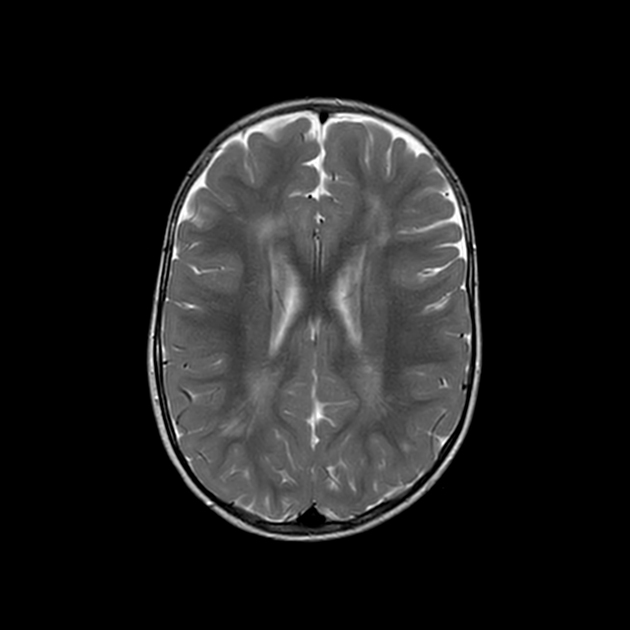
On T2-weighted sequences, there is high signal in the white matter, especially in the frontal and temporal lobes surrounding the ventricular horns 1. Temporal lobe cysts-like spaces may also form 1.
Over time, atrophy develops, particularly of the periventricular regions 1. The brainstem and cerebellum may also develop prominent atrophy 1.
Arteriopathy is also a prominent feature of SAMHD1 mutations with aneurysms, stenoses and moyamoya pattern encountered 1.
Treatment and prognosis
Although no established treatment exists, there is evidence that immune modulation (e.g. corticosteroids) during the active phase of the disease may be of benefit 1. Unfortunately, judging the efficacy of these interventions is difficult10. In addition, the use of JAK inhibitors like baricitinib have been shown to block the downstream effects of interferon activation and improve symptoms 6,7. Clinical trials on the long-term efficacy and safety of JAK inhibitors in AGS patients are ongoing.
Although not established beyond doubt, it appears that Aicardi-Goutières syndrome represents a monophasic encephalopathy in infancy without ongoing progression. Death is therefore believed to be the consequence of neurological damage that occurred during the active phase of the disease rather than representing true disease progression.
History and etymology
The condition was named after the French pediatric neurologists Jean Aicardi (1926-2015) 4 and Francoise Goutières (fl. 2024) who described the first case in 1984.
Differential diagnoses
Although many other leukodystrophies are viable differential diagnoses, additional conditions to be considered include 1:
antenatal/perinatal infections (e.g. TORCH infections)
band-like calcification polymicrogyria (BLC-PMG; pseudo-TORCH syndrome)
classic Cockayne syndrome (CS type 1)
cerebroretinal microangiopathy with calcifications and cysts (CRMCC; Coats plus)
mitochondrial cytopathies (e.g. Leigh syndrome)


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.