
Hurler or Hurler-Scheie syndrome is one of the mucopolysaccharidoses (MPS type I).
On this page:
Epidemiology
The estimated incidence is ~1:100,000.
Diagnosis
The diagnosis may be suspected with characteristic skeletal, ophthalmic, neurodevelopmental, or biochemical findings. Urine analysis may show mucopolysaccharides 4. The diagnosis is made by demonstrating α-l-iduronidase activity deficiency and raised glycosaminoglycan levels, or molecular testing showing a pathogenic α-L-iduronidase mutation 4,5.
Clinical presentation
Hurler syndrome manifests in the first years of life with intellectual disability, corneal clouding, deafness, and cardiac disease. Death usually occurs within the first decade of life, often from cardiac disease.
Pathology
Hurler syndrome is a lysosomal storage disorder from abnormal mucopolysaccharide metabolism due to deficient α-l-iduronidase activity responsible for the hydrolysis of glycosaminoglycans 4.
Genetics
Inheritance is autosomal recessive 4.
Radiographic features
-
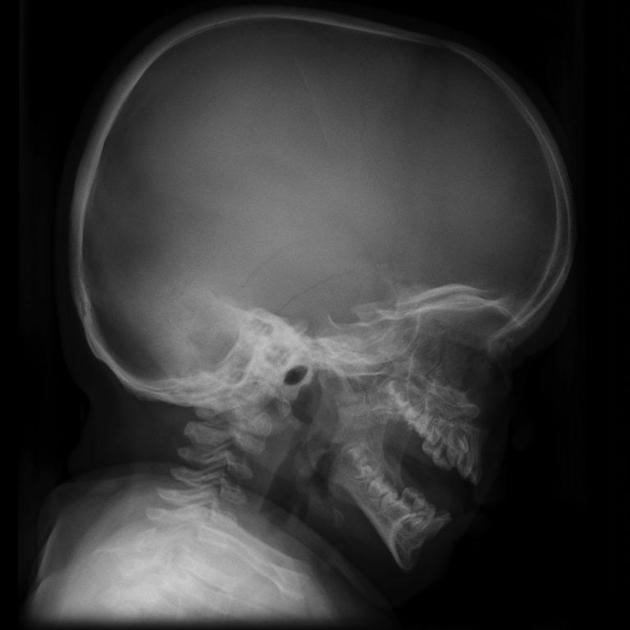
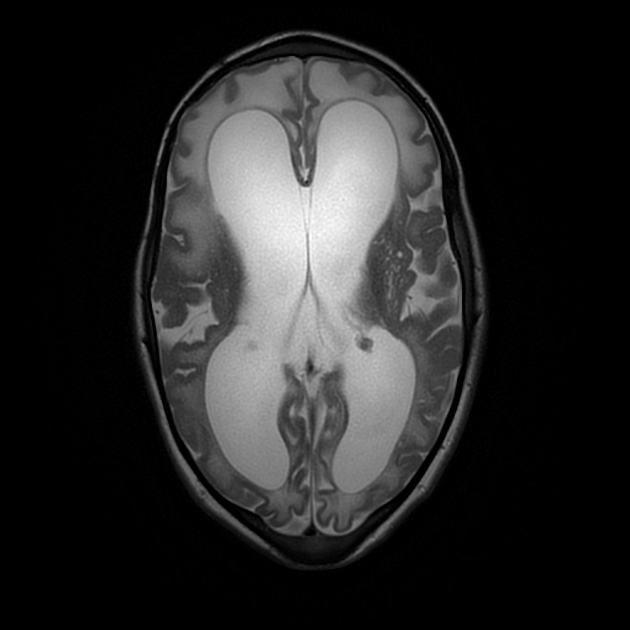
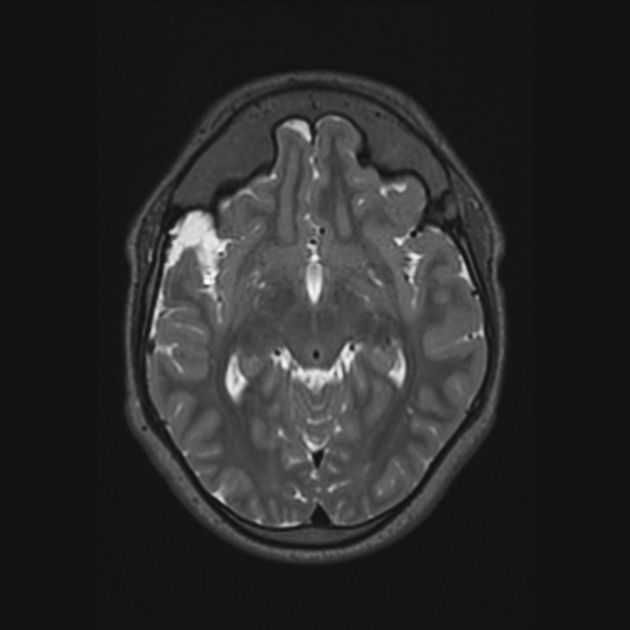
neurological anomalies
prominent perivascular spaces
diffuse white matter changes
pachymeningiopathy
-
cervicomedullary junction anomalies
-
cord compression at the craniovertebral junction:
C1-C2 subluxation: atlantoaxial subluxation
narrowing of the foramen magnum due to a combination of short C1 arch, dysplastic odontoid, and thickened meninges and ligaments
-
-
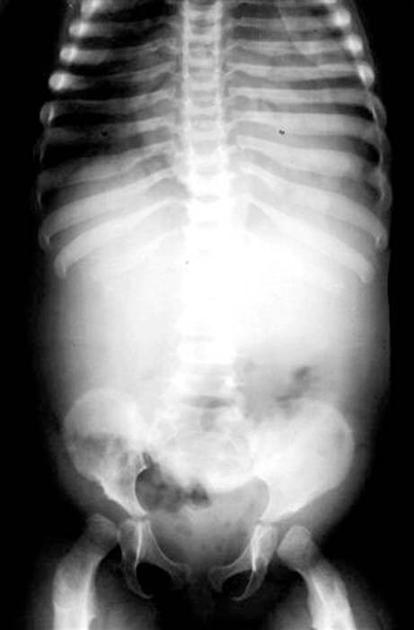
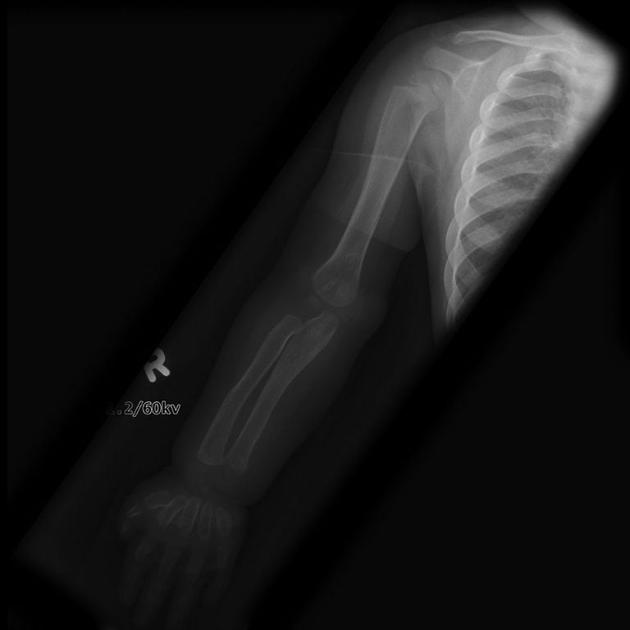
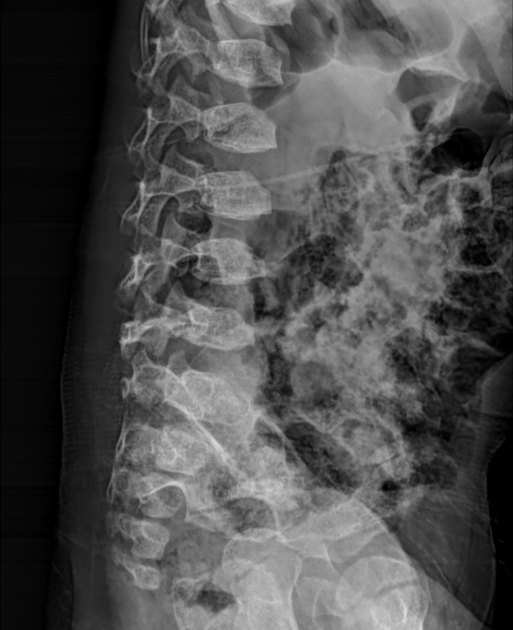
skeletal anomalies
concave articular surface of the mandibular condyle
shortening and widening of long bones
left pointing of proximal metacarpals
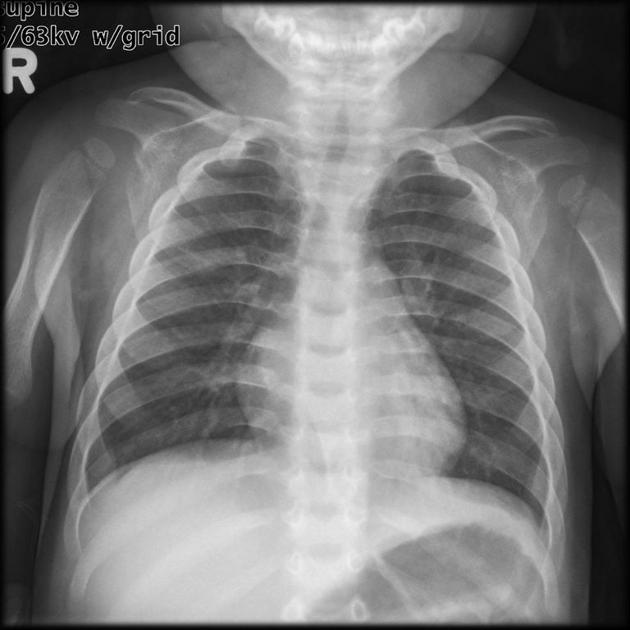
widening of anterior ribs (oar-shaped / paddle ribs) and clavicles
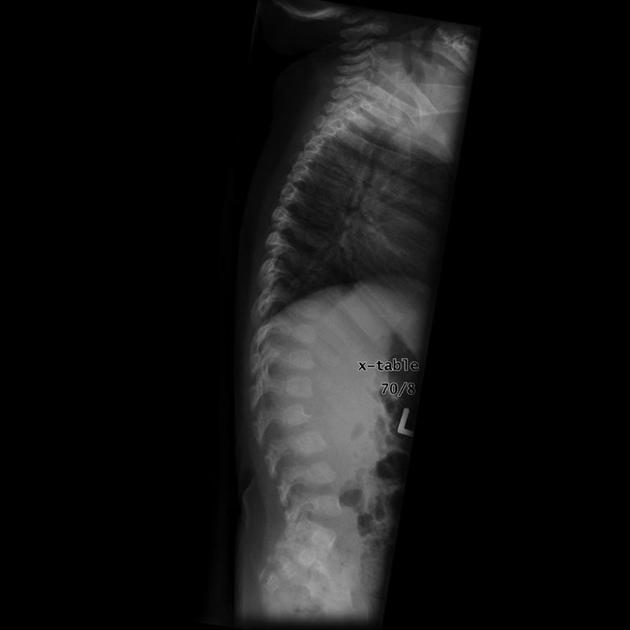
thoracolumbar kyphosis or hypoplastic vertebra at thoracolumbar junction results in gibbus deformity
-
heart involvement/anomalies
cardiac valve disease: early-onset severe regurgitation and stenosis
coronary artery disease
cardiomegaly: initially hypertrophic then dilated
-
other features
History and etymology
It is named after Gertrud Hurler (1889-1965), a German pediatrician 1.


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.