
Krabbe disease, also known as globoid cell leukodystrophy, is an autosomal recessive lysosomal storage disorder resulting in damage to cells involved in myelin turnover. It thus affects both the peripheral nervous system and the central nervous system (manifesting as a leukodystrophy).
On this page:
Epidemiology
The majority of individuals with Krabbe disease present in early childhood although adult presentations as late as the 5th decade are encountered 9.
Krabbe disease can be divided according to the age of onset into two or three forms depending on the author 2,3,5:
infantile form (85-90%): rapidly progressive usual onset <2 years of age
late-onset/adult form (10-15%): more slowly progressive
Clinical presentation
Clinical presentation can vary and depends on the age of onset with numerous clinical features including 5,9:
hypertonia
irritability
delayed milestones
loss of developed milestones
myoclonus
nystagmus
bulbar palsies
blindness
cognitive decline
Pathology
Genetics
Krabbe disease is caused by mutations in the GALC gene (mapped to chromosome 14q) which encodes galactocerebrosidase, an enzyme that degrades galactosylceramide, a normal constituent of myelin. Deficiency of galactocerebrosidase results in the accumulation of galactosylceramide within the lysosomes of Schwann cells and oligodendrocytes which eventually results in apoptosis with secondary abnormal activation of microglia and macrophages with subsequent demyelination and gliosis 2,9,10. The multinucleated macrophages are known as globoid cells 10.
Radiographic features
Changes typically involve the corticospinal tracts 13.
CT
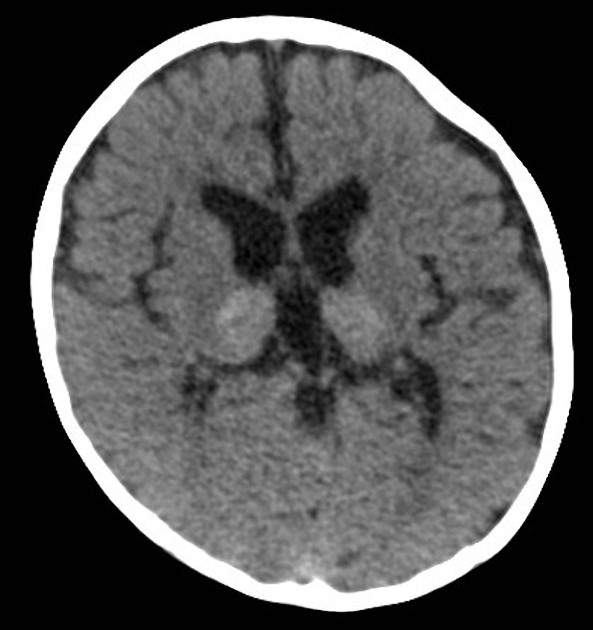
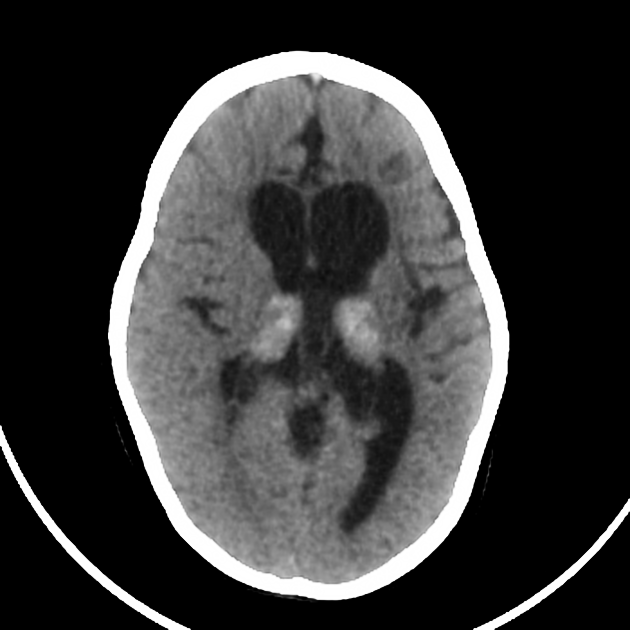
Initially, CT may show hyperdense areas symmetrically involving the thalami 1, cerebellum, caudate nuclei, posterior limbs of the internal capsule, and brainstem. Changes may also extend into the centrum semiovale/corona radiata region 4,5.
Later, hypoattenuation develops in the white matter of the centrum semiovale 2. Eventually, atrophy of the cerebellum and cerebrum develops 2.
MRI
When diffuse white matter involvement develops, MRI features are characterized by:
-
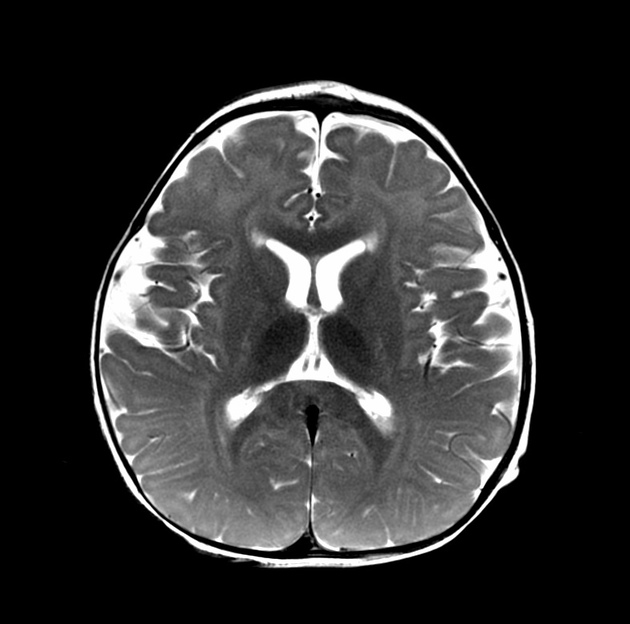
T2
high signal involving parieto-occipital predominant or diffuse periventricular white matter 2 involvement and predilection for corticospinal tracts including through the internal capsules and brainstem 9,13
subcortical U-fibers may be spared until late in the course of the disease 5
a tigroid pattern of white matter involvement can be seen 12
T1 C+ (Gd): no contrast enhancement in these areas 2
MRS: adult form may show abnormal choline elevation in centrum semiovale 7
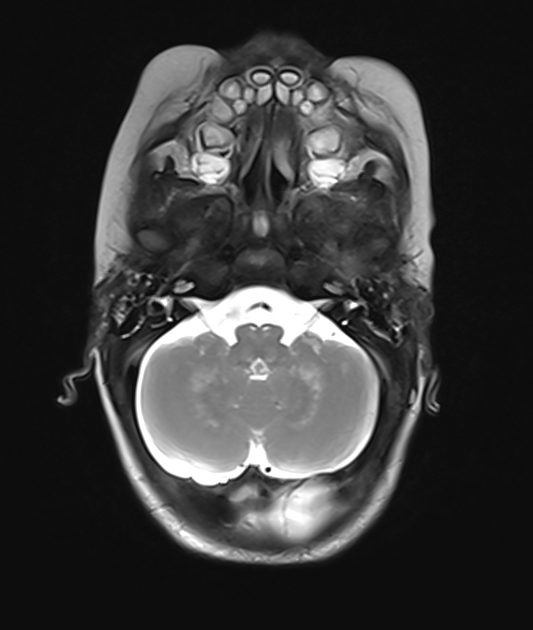
Enhancement and enlargement of the optic nerves and occasionally peripheral nerves (e.g. lumbosacral plexus) can also be seen 2,12.
Treatment and prognosis
Therapy primarily centers on hematopoietic stem cell transplantation which can delay disease progression 11.
Prognosis depends to a large degree on the age at which a diagnosis is made. Infantile-onset represents a more aggressive course, presenting early and leading to a more rapid neurological decline.
History and etymology
It is named after Knud Haraldsen Krabbe, a Danish neurologist (1885-1961) 6.
Differential diagnosis
The differential diagnosis is largely that of other leukodystrophies, mainly metachromatic leukodystrophy 12. Neonatal hypoxic-ischemic encephalopathy can also occasionally present with similar imaging findings. In adult-onset forms, amyotrophic lateral sclerosis may be a clinicoradiological differential diagnosis 13.


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.