
Central neurocytoma
Updates to Article Attributes
Central neurocytomas are WHO Grade II neuroepithelial intraventricular tumours with fairly characteristic imaging features, appearing as heterogeneous masses of variable size and enhancement within the lateral ventricle. They are typically seen in young patients, and generally have a good prognosis provided a complete resection can be achieved.
Epidemiology
Central neurocytomas are typically seen in young patients (20-40 years of age), and accounts for less than 1% (0.25-0.5%) of intracranial tumours. There is no reported gender predilection 10.
Clinical presentation
Typically central neurocytomas present with symptoms of increased intracranial pressure, headaches being most frequent, or seizures (especially tumours with extra ventricular extension).
A relatively short clinical course, typically only a few months, is most common. Rarely central neurocytomas may be associated with sudden death secondary to acute ventricular obstruction 4. Also rare, is a sudden presentation due to intraventricular haemorrhage 7.
Pathology
Initially described as recently as 1982, central neurocytomas demonstrate neuronal differentiation and histologically appear similar to oligodendrogliomas. This has historically has resulted in many tumours erroneously categorised. They lack co-deletioncodeletion of 1p19q which is characteristic of oligodendroglioma. The cells are typically uniform and round with a salt and pepper appearance.
The initial description classified them as WHO grade I lesions, however this was upgraded in 1993 to WHO grade II as it was recognised that at least some of these tumours exhibited more aggressive behaviour 10.
Markers
Purely neuronal origin is demonstrated positivity to neuronal markers such as:
- synaptophysin
- neuronal specific enolase
Variants
Ganglioneurocytoma: shows differentiation towards ganglion cells 6.
Radiographic features
The vast majority of central neurocytomas are located entirely within the ventricles. Typical locations include 4.
- lateral ventricles around foramen of Monro (most common): 50%
- both lateral and 3rd ventricles: 15%
- bilateral: 15%
- 3rd ventricle in isolation: 5%
Extra ventricular neurocytomas (or cerebral neurocytomas) are distinctly uncommon, and thought to be a separate entity due to the tendency to have prominent ganglionic or glial differentiation.
CT
Central neurocytomas are usually hyperattenuating compared to white matter. Calcification seen in over half of cases, usually punctate in nature 4,10. Cystic regions are frequently present, especially in larger tumours. Contrast enhancement is usually mild to moderate. Accompanying ventricular dilatation often present.
MRI
-
T1
- isointense to grey matter
- heterogenous
-
T1 C+
- mild-moderate heterogeneous enhancement
-
T2/FLAIR
- typically iso to somewhat hyperintense compared to brain
- numerous cystic areas (bubbly appearance), many of which completely attenuate on FLAIR
- prominent flow voids may be seen 10
-
GE/SWI
- calcification is common, typically punctate
- haemorrhage (especially in larger tumours) is common
- uncommonly results in ventricular haemorrhage
-
MR spectroscopy
- may have a strong choline peak
- glycine peak (3.55ppm) has also been reported 10
Angiography
A tumour blush is frequently identified, with the mass supplied by choroidal vessels. No large feeding arteries are usually seen.
Treatment and prognosis
Complete surgical resection is usually curative (5 years survival 81%). When only incomplete resection possible or extraventricular extension is present then adjuvant radiotherapy (and sometimes chemotherapy) are added, although their benefit is not well established.
Cases of CSF dissemination have been reported, but are rare 10.
Differential diagnosis
-
ependymoma
- more frequent in childhood
- more commonly in 4th ventricle
- supratentorial tumours (esp in children) often have a significant extraventricular (parenchymal) component 4
-
intraventricular meningioma
- homogeneous contrast enhancement
- well circumscribed mass
-
subependymoma
- typically found in the 4th ventricle
- usually older individuals 8
- may have ependymoma components and look very similar 9
-
subependymal giant cell astrocytoma (SGCA)
- in patients with tuberous sclerosis
- vivid contrast enhancement
-
choroid plexus papilloma (CPP)
- mainly in children
- typically show intense contrast enhancement
-
intra ventricular metastasis
- older patients
- usually stronger contrast enhancement
- history of primary (e.g. RCC)
-
oligodendroglioma
- this is especially difficult in cases where there is a parenchymal component as histologically the tumours are very similar
-<p><strong>Central neurocytomas</strong> are <a href="/articles/cns-tumours-classification-and-grading-who">WHO Grade II</a> neuroepithelial <a href="/articles/intraventricular-neoplasms-and-lesions">intraventricular tumours</a> with fairly characteristic imaging features, appearing as heterogeneous masses of variable size and enhancement within the lateral ventricle. They are typically seen in young patients, and generally have a good prognosis provided a complete resection can be achieved. </p><h4>Epidemiology</h4><p>Central neurocytomas are typically seen in young patients (20-40 years of age), and accounts for less than 1% (0.25-0.5%) of <a href="/articles/brain-tumours">intracranial tumours</a>. There is no reported gender predilection <sup>10</sup>. </p><h4>Clinical presentation</h4><p>Typically central neurocytomas present with symptoms of increased intracranial pressure, headaches being most frequent, or seizures (especially tumours with extra ventricular extension).</p><p>A relatively short clinical course, typically only a few months, is most common. Rarely central neurocytomas may be associated with sudden death secondary to acute ventricular obstruction <sup>4</sup>. Also rare, is a sudden presentation due to <a href="/articles/intraventricular-haemorrhage">intraventricular haemorrhage</a> <sup>7</sup>. </p><h4>Pathology</h4><p>Initially described as recently as 1982, central neurocytomas demonstrate neuronal differentiation and histologically appear similar to <a href="/articles/oligodendroglioma">oligodendrogliomas</a>. This has historically has resulted in many tumours erroneously categorised. They lack co-deletion of 1p19q which is characteristic of oligodendroglioma. The cells are typically uniform and round with a salt and pepper appearance.</p><p>The initial description classified them as WHO grade I lesions, however this was upgraded in 1993 to WHO grade II as it was recognised that at least some of these tumours exhibited more aggressive behaviour <sup>10</sup>. </p><h5>Markers</h5><p>Purely neuronal origin is demonstrated positivity to neuronal markers such as:</p><ul>- +<p><strong>Central neurocytomas</strong> are <a href="/articles/cns-tumours-classification-and-grading-who">WHO Grade II</a> neuroepithelial <a href="/articles/intraventricular-neoplasms-and-lesions">intraventricular tumours</a> with fairly characteristic imaging features, appearing as heterogeneous masses of variable size and enhancement within the lateral ventricle. They are typically seen in young patients, and generally have a good prognosis provided a complete resection can be achieved. </p><h4>Epidemiology</h4><p>Central neurocytomas are typically seen in young patients (20-40 years of age), and accounts for less than 1% (0.25-0.5%) of <a href="/articles/brain-tumours">intracranial tumours</a>. There is no reported gender predilection <sup>10</sup>. </p><h4>Clinical presentation</h4><p>Typically central neurocytomas present with symptoms of increased intracranial pressure, headaches being most frequent, or seizures (especially tumours with extra ventricular extension).</p><p>A relatively short clinical course, typically only a few months, is most common. Rarely central neurocytomas may be associated with sudden death secondary to acute ventricular obstruction <sup>4</sup>. Also rare, is a sudden presentation due to <a href="/articles/intraventricular-haemorrhage">intraventricular haemorrhage</a> <sup>7</sup>. </p><h4>Pathology</h4><p>Initially described as recently as 1982, central neurocytomas demonstrate neuronal differentiation and histologically appear similar to <a href="/articles/oligodendroglioma">oligodendrogliomas</a>. This has historically has resulted in many tumours erroneously categorised. They lack <a title="1p19q codeletion" href="/articles/1p19q-codeletion">codeletion of 1p19q</a> which is characteristic of oligodendroglioma. The cells are typically uniform and round with a salt and pepper appearance.</p><p>The initial description classified them as WHO grade I lesions, however this was upgraded in 1993 to WHO grade II as it was recognised that at least some of these tumours exhibited more aggressive behaviour <sup>10</sup>. </p><h5>Markers</h5><p>Purely neuronal origin is demonstrated positivity to neuronal markers such as:</p><ul>
Image 3 Pathology ( create )

Image 4 MRI (FLAIR) ( update )

Image 5 MRI (T2) ( update )
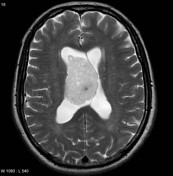
Image 6 MRI (T2) ( update )

Image 7 MRI (T1 C+ fat sat) ( update )

Image 8 MRI (T2) ( update )

Image 9 MRI (T1 C+) ( update )

Image 10 MRI (T2) ( update )

Image 11 MRI (Gradient Echo) ( update )
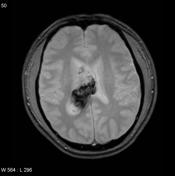
Image 12 MRI (T2) ( update )

Image 13 MRI (T2) ( update )
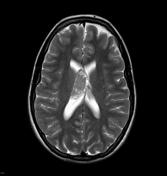
Image 18 MRI (T1 C+) ( update )

Image 19 MRI (T2) ( create )

Image 21 MRI (T2) ( update )






 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.