
Pancreatic neuroendocrine tumors (pNET), also known as endocrine tumors of the pancreas, arise from pancreatic ductal stem cells and include some distinct tumors that match the cell type of origin.
On this page:
Terminology
Pancreatic neuroendocrine tumors have commonly been referred to as "islet cell tumors", referring to the islets of Langerhans, from which they were thought to derive. It has since been shown that these tumors derive from ductal pluripotent stem cells, and "neuroendocrine tumor" is now preferred 3.
Epidemiology
Overall, pancreatic neuroendocrine tumors have an incidence of 0.001% and account for 1-2% of pancreatic neoplasms. They occur most commonly at ages 30-60 with no clear gender predilection.
Associations
Most tumors are sporadic. Approximately 10% are associated with multiple endocrine neoplasia type 1 (MEN1), von Hippel Lindau disease, tuberous sclerosis, and neurofibromatosis type 1 7.
Clinical presentation
Syndromic tumors tend to present earlier, with clinical signs and symptoms related to their cell type and biological activity:
non-functioning tumors: tend to present late and often larger in size
Pathology
Neuroendocrine tumors are classically defined by the expression of markers of neuroendocrine differentiation (including chromogranin A and synaptophysin) and hormone production.
These tumors can broadly be divided according to whether or not they secrete enough active compounds to be functional or not:
non-functional tumors: account for ~67.5% (range 50–85%) of PNETs 7
-
functional tumors
VIPoma: rare ref
somatostatinoma: rare, some of these can be non-functional
The term "syndromic" may be preferred over "functioning" since it is becoming increasingly clear that most tumors are functional (i.e. produce hormones), but either do not produce enough hormone or produce an ineffective form of the hormone, so that they may not produce a clinical syndrome ref.
Biological behavior also depends on the cell of origin ref:
insulinoma: 10% malignant
gastrinoma: 60% malignant
glucagonoma: 80% malignant
VIPoma: 75% malignant
somatostatinoma: 75% malignant
non-functional: 85-100% malignant
Classification
According to the 2017 World Health Organization (WHO) classification, these tumors are histologically graded as 5,6:
-
well-differentiated status
grade 1 (G1): tumor expressing <2 mitoses/2 mm2 and ≤2% Ki-67 index
grade 2 (G2): tumor expressing between 2 to 20 mitoses/2 mm2 and 3 to 20% Ki-67 index
grade 3 (G3): more than 20 mitoses/2 mm2 and more than 20% Ki-67 index
-
poorly-differentiated and containing components of adenocarcinoma - they are named pancreatic neuroendocrine carcinoma (pNEC) and divided into two types
small cell type
large cell type
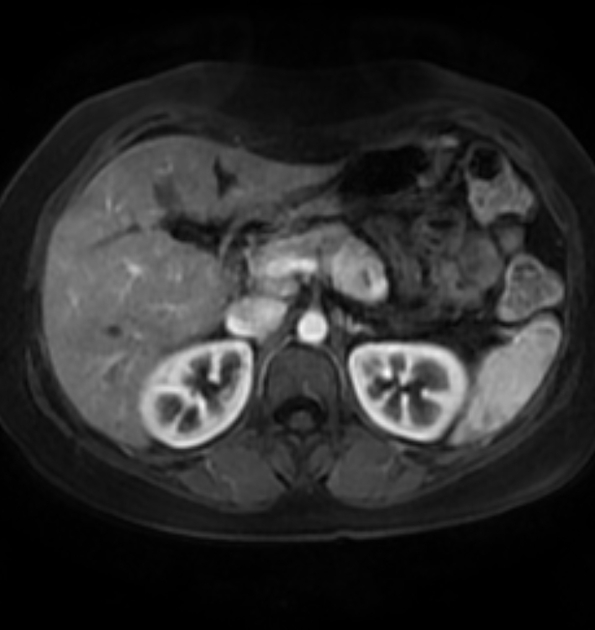
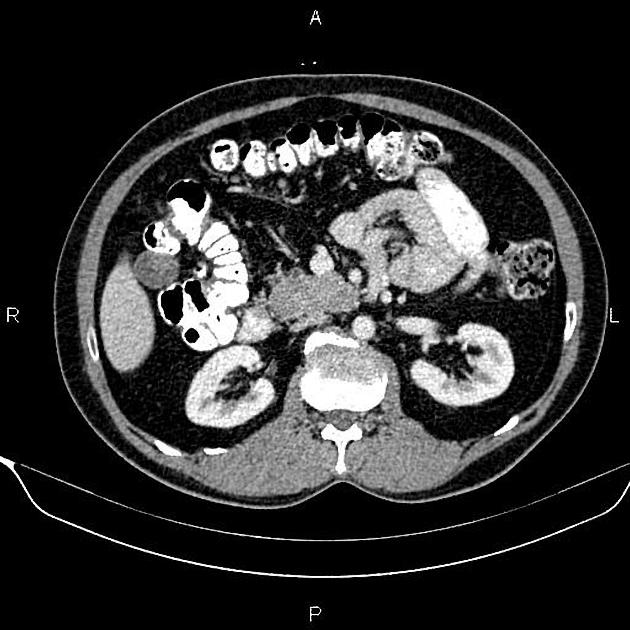
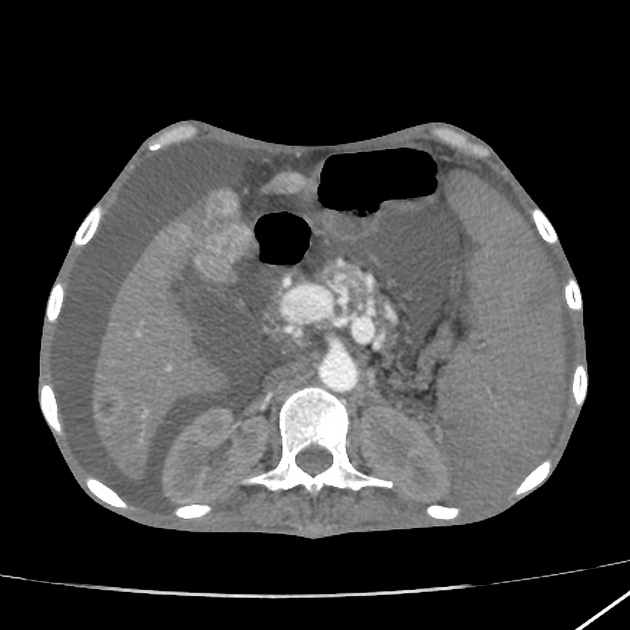
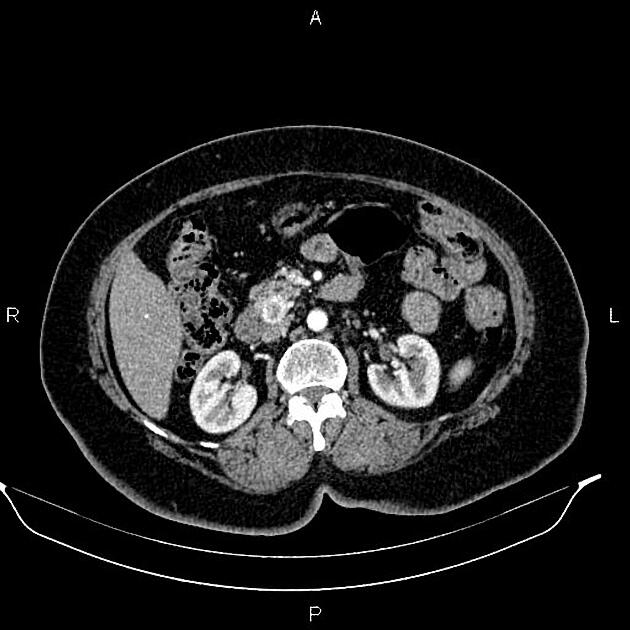
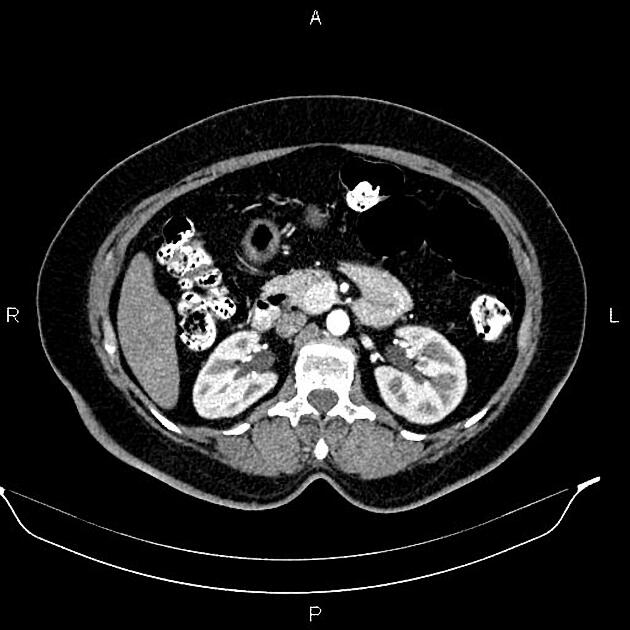
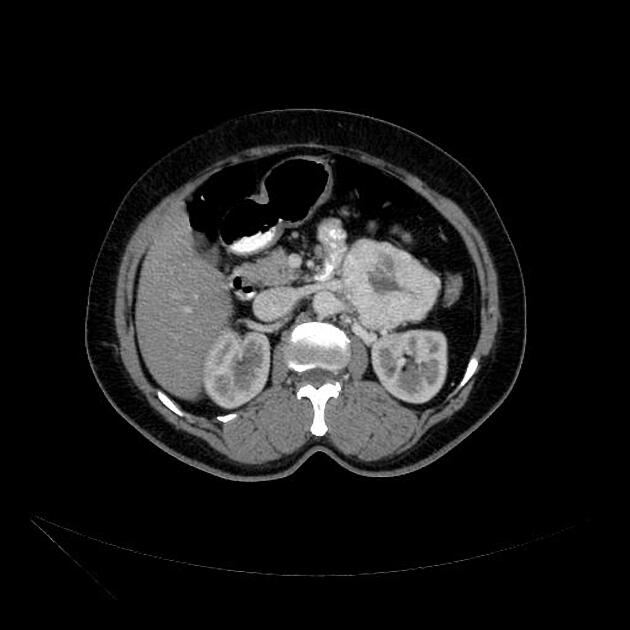
Radiographic features
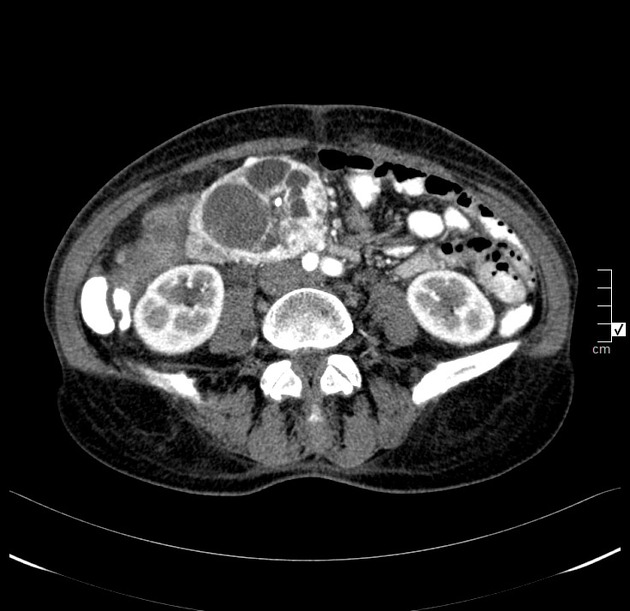
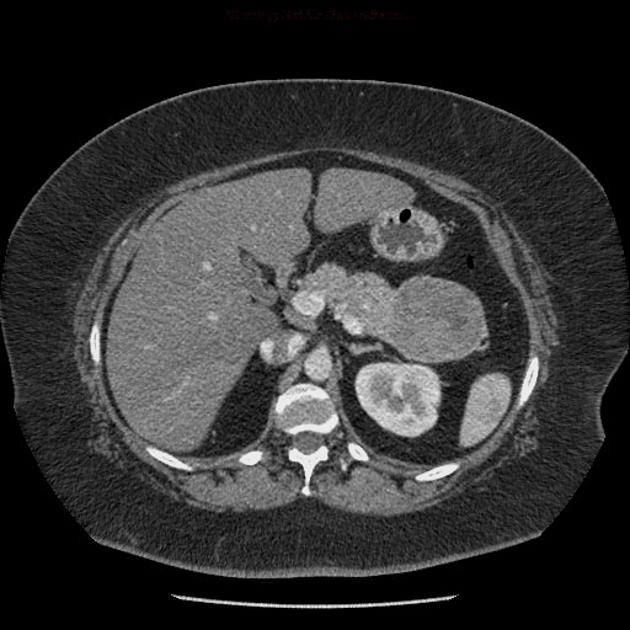
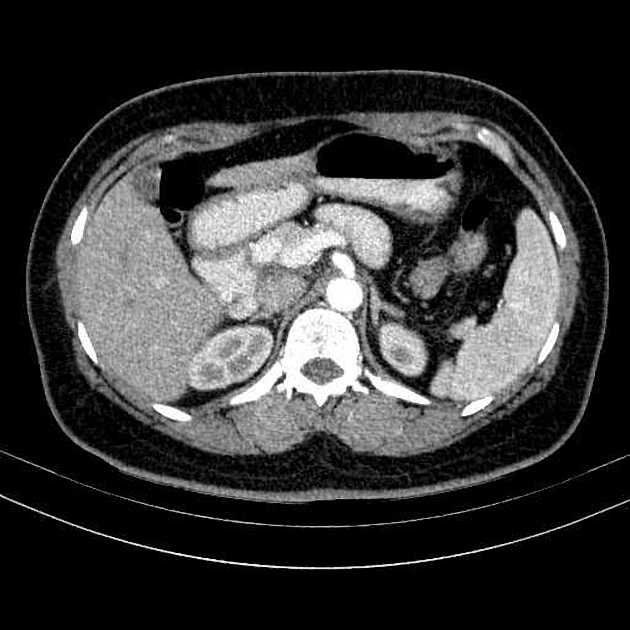
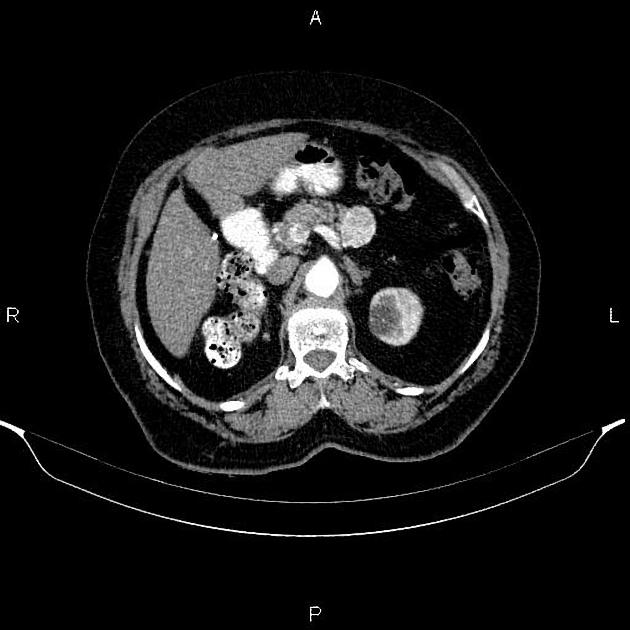
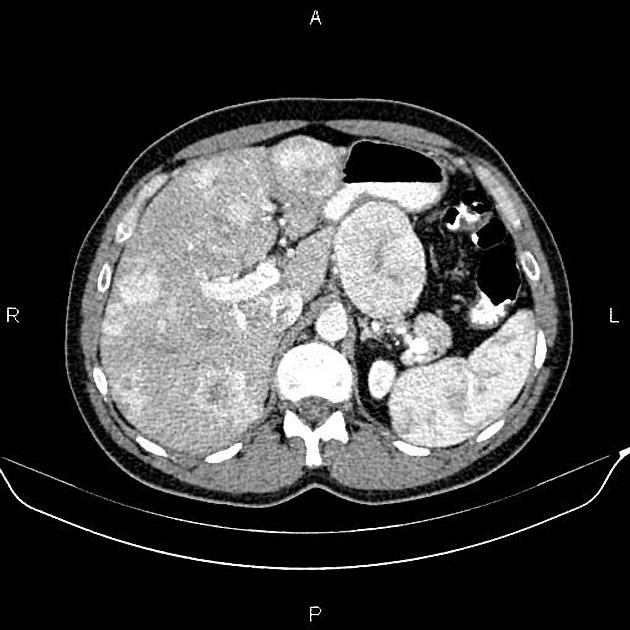
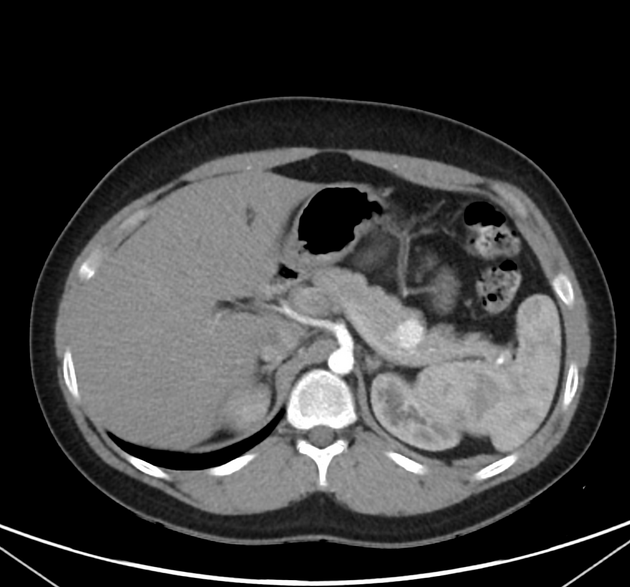
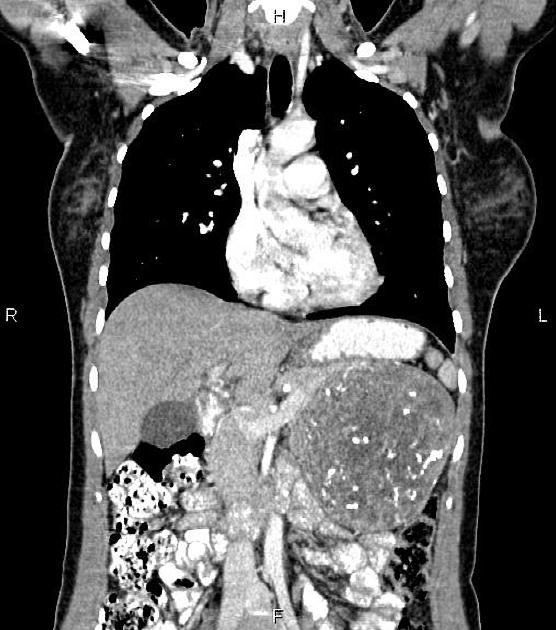
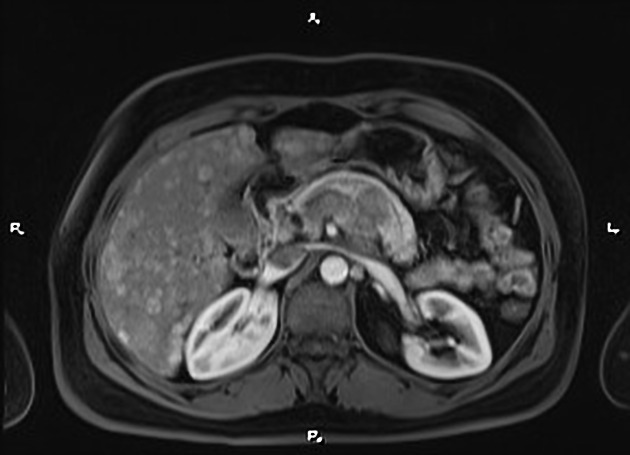
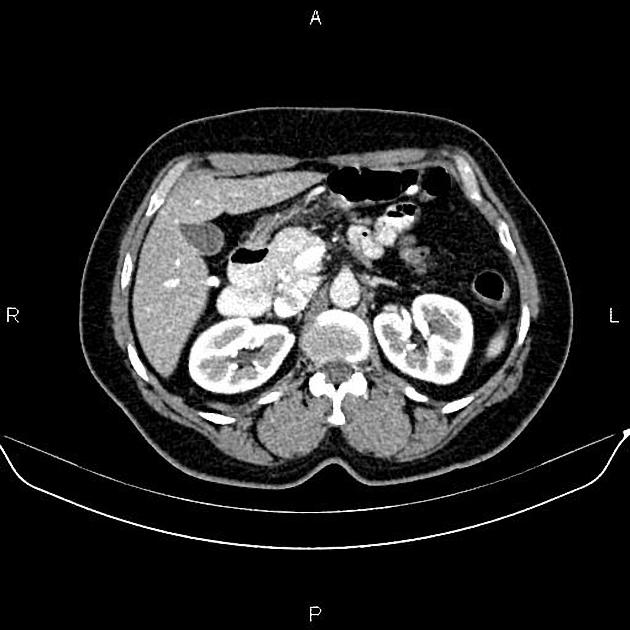
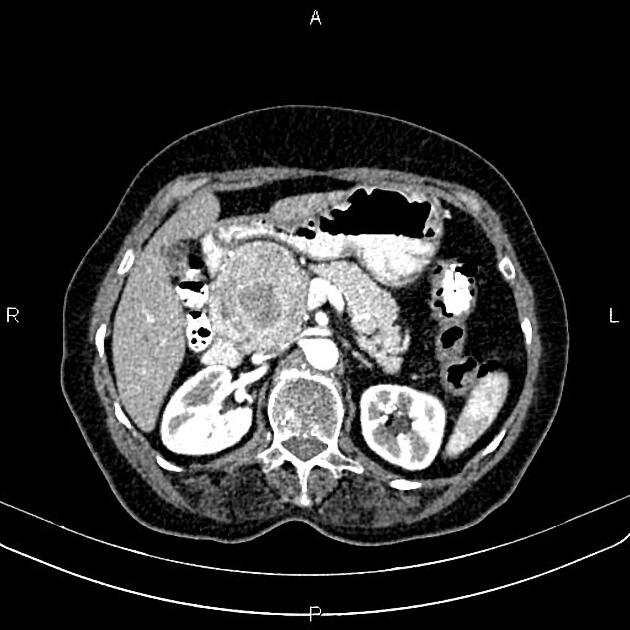
Overall these neuroendocrine tumors of the pancreas tend to be highly vascular and well-circumscribed, often displacing adjacent structures. They can demonstrate calcific or cystic change.
Ultrasound
well-circumscribed with smooth margins
round or oval
hypoechoic
Liver metastases may be hyperechoic or targetoid.
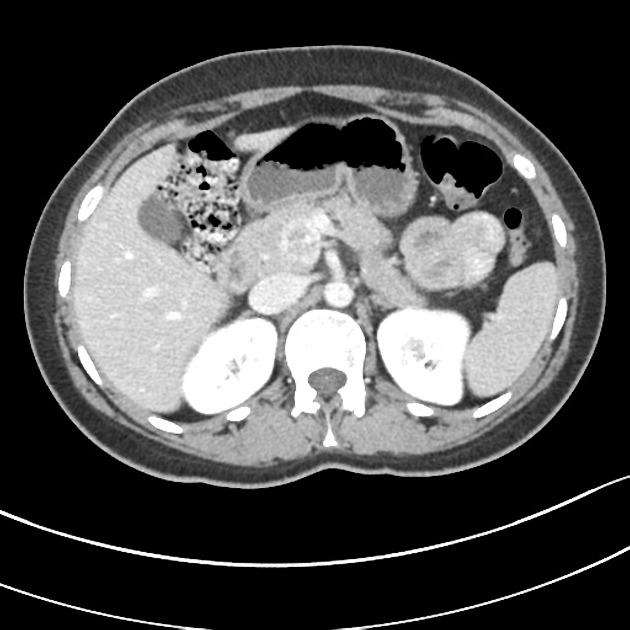
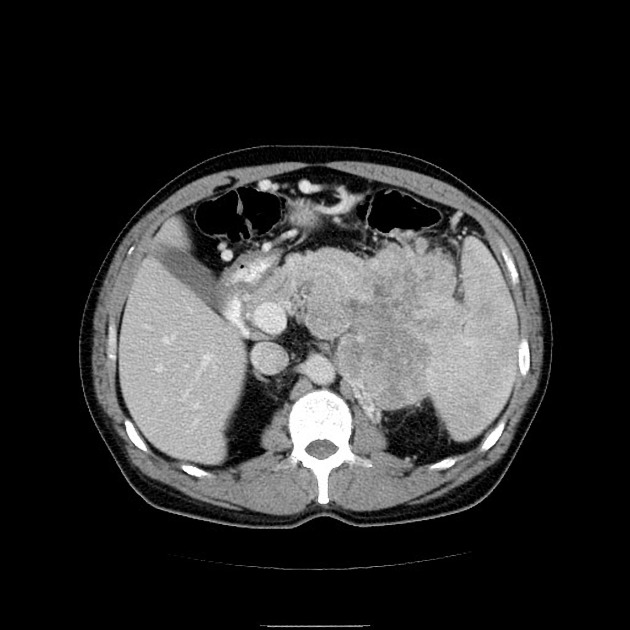
CT
Smaller tumors:
hypervascular
tend to be homogenous and well-circumscribed
Larger tumors:
may appear heterogeneous and contain areas of cystic or necrotic change
can occasionally manifest as primarily cystic lesions and are distinguishable from other cystic neoplasms by their hypervascular rim
Since pNETs usually have a distinct capsule, this means that they displace rather than invade surrounding structures as they grow in size. As a result, they less frequently present with biliary obstruction, which is a classic mode of presentation for pancreatic adenocarcinomas.
Neuroendocrine tumors of the pancreas show peak contrast enhancement in the early arterial phase (25-35 s) rather than in the late arterial phase (35-45 s) which is normally used for pancreatic imaging. This is particularly important when considering that small lesions may be missed in the late arterial phase when the tumor will appear isointense with enhancing pancreatic parenchyma. Accessory spleens or splenules can mimick small neuroendocorine tumors. However, the washout pattern of splenules mirrors that of the adjacent spleen 8.
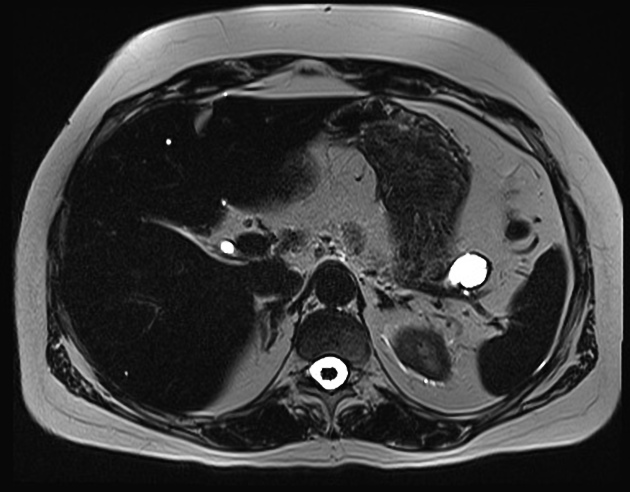
MRI
Sensitivity is similar to CT
T1: hypointense relative to pancreas
T2: typically hyperintense relative to the pancreas, but there is a range of signal intensities
T1 C+ (Gd): hyperintense/hypervascular relative to pancreas
DWI/ADC: restricted diffusion is usually present and tends to correlate to the degree of tumor differentiation
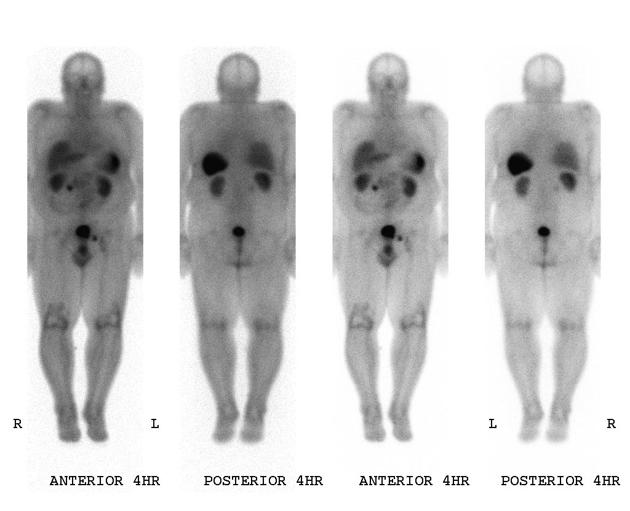
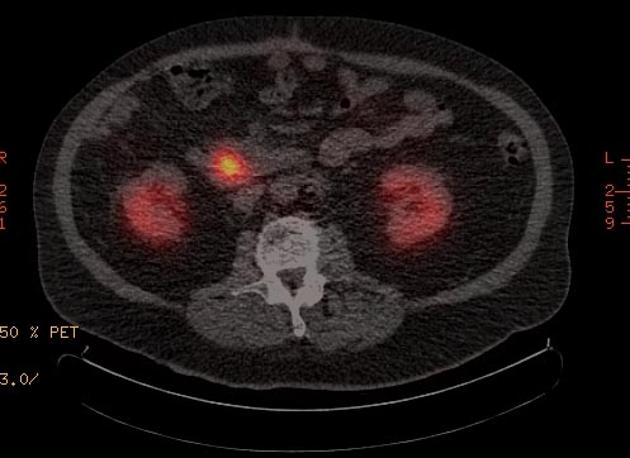
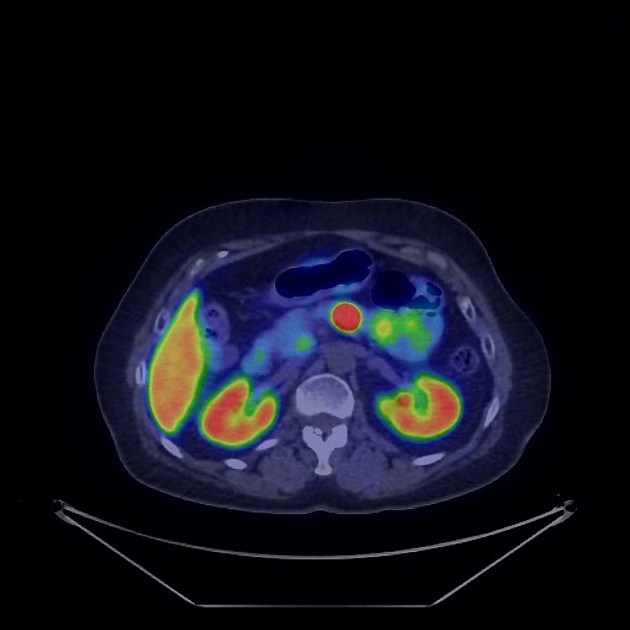
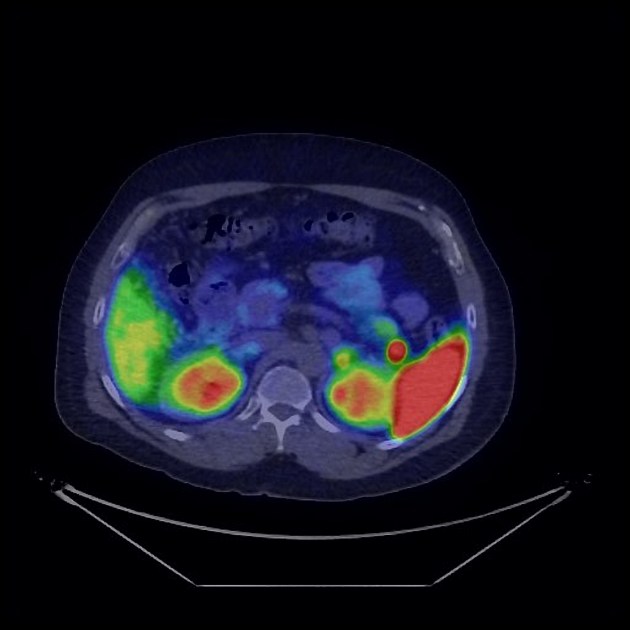
Nuclear medicine
Nuclear medicine studies play an important role in the staging of neuroendocrine tumors.
-
gallium-68 DOTATATE (or DOTATOC, or DOTANOC) PET-CT
superior to octreotide scan
-
F-18 FDG PET-CT
sensitivity limited unless poorly-differentiated
-
planar or SPECT
-
sensitivity is ~80%, although limited by the somatostatin receptor characteristics of the tumor
reported sensitivity is highest with gastrinomas >2 cm
reported sensitivity is lowest with insulinomas
has been largely replaced by PET-CT in most centers
Treatment and prognosis
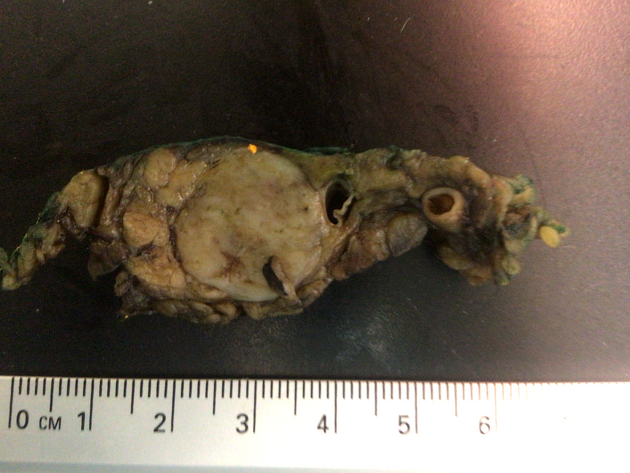
Surgical resection for low-grade localized PNETs can be curative and should be considered for all symptomatic/functional PNETs and non-function PNETs >2 cm in size 7. Non-functional PNETs may be suitable for active surveillance if <1-2 cm in size 7.
Even patients with advanced disease can have reasonable long-term survival ref. Recurrence is associated with 7:
tumor size >2 cm
symptomatic tumors
Ki67 >3%
nodal disease
Differential diagnosis
metastasis (e.g. renal cell carcinoma)
intrapancreatic splenule (if in the tail of the pancreas)
mostly-solid serous cystadenoma
Practical points
check for concomitant metastatic disease: neuroendocrine tumors most commonly give metastases in the liver and, less frequently, in the bones


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.