
Pulmonary manifestations of scleroderma are demonstrated histologically in 90% of patients with scleroderma. It is a leading cause of mortality and at autopsy the lung is reportedly involved in close to 100% of cases. However, only 25% of patients will present with respiratory symptoms or demonstrate abnormalities on CT 4.
For a discussion of scleroderma in general, refer to parent article: scleroderma.
On this page:
Clinical presentation
In patients who are symptomatic due to pulmonary involvement, respiratory function tests typically demonstrate a restriction pattern, consisting of 4:
diminished lung volumes
preserved flow rates
low diffusion capacity
Pathology
The pathogenesis of pulmonary involvement relates to separate mechanisms:
-
direct involvement
-
scleroderma-related interstitial lung disease (SSc-ILD)
non-specific interstitial pneumonitis (NSIP): more common
usual interstitial pneumonia (UIP) pattern: histologically indistinguishable from rheumatoid lung and idiopathic pulmonary fibrosis
-
scleroderma-related pulmonary arterial hypertension (SSc-PAH)
pathogenesis is unclear but may be due to vascular endothelial injury with subsequent apoptosis, inflammation and dysregulated angiogenesis leading to arterial obliteration and narrowing from fibrosis 6
-
-
indirect involvement
secondary lung injury due to aspiration pneumonitis secondary to esophageal involvement: see gastrointestinal manifestations of scleroderma
The two pathologies may co-exist. The remainder of the article discusses the former. Please refer to aspiration pneumonitis for discussion of that entity.
Radiographic features
Plain radiograph
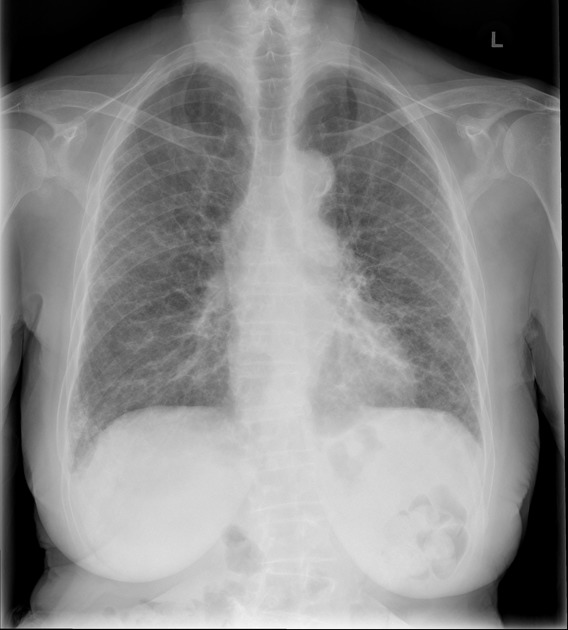
Chest radiographs are insensitive to early changes and may appear normal despite respiratory function test abnormalities. Eventually, changes of pulmonary fibrosis become evident. Additional features include 4:
dilated esophagus
eggshell calcification of mediastinal nodes
pleural effusions are uncommon
enlargement of the cardiac silhouette and pulmonary arteries due to scleroderma induced pulmonary vascular disease may also be evident.
CT
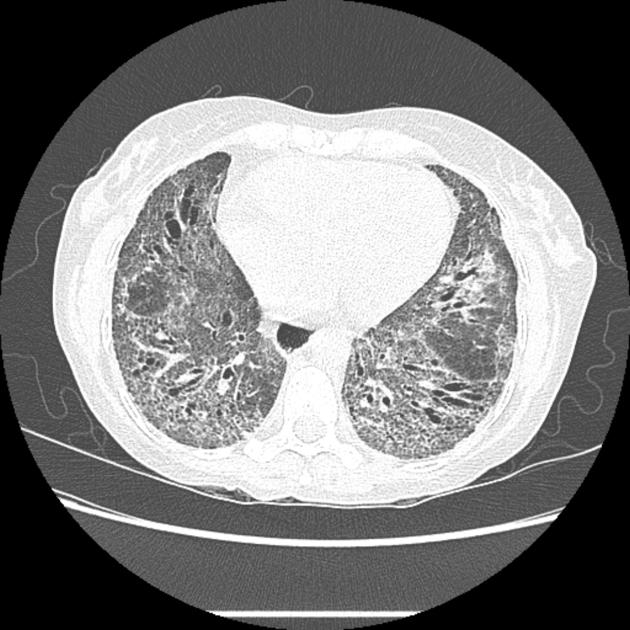
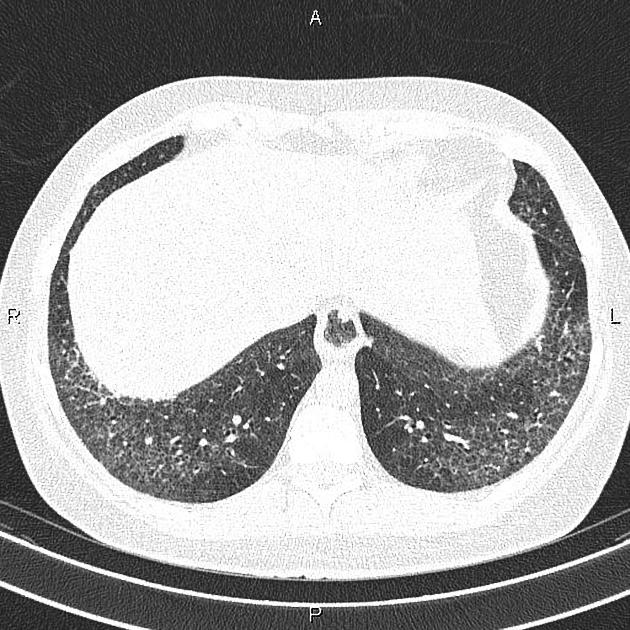
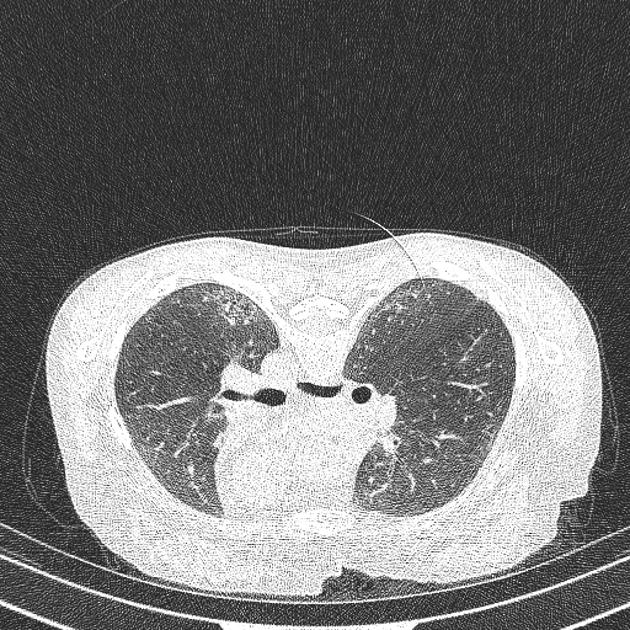
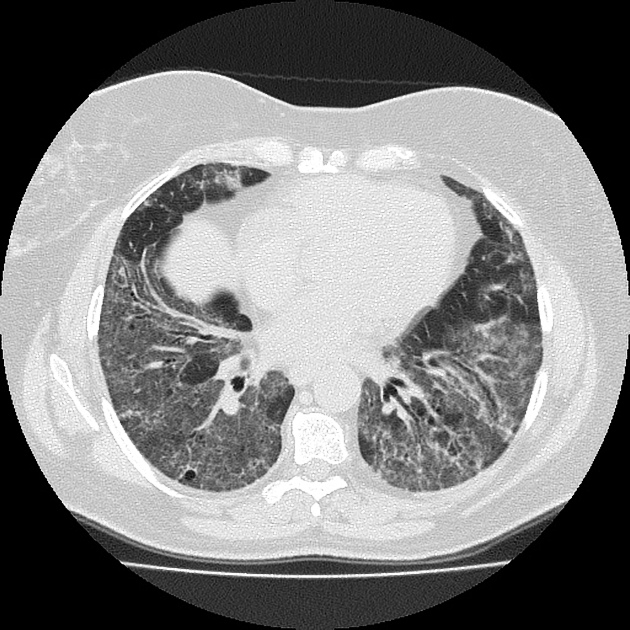
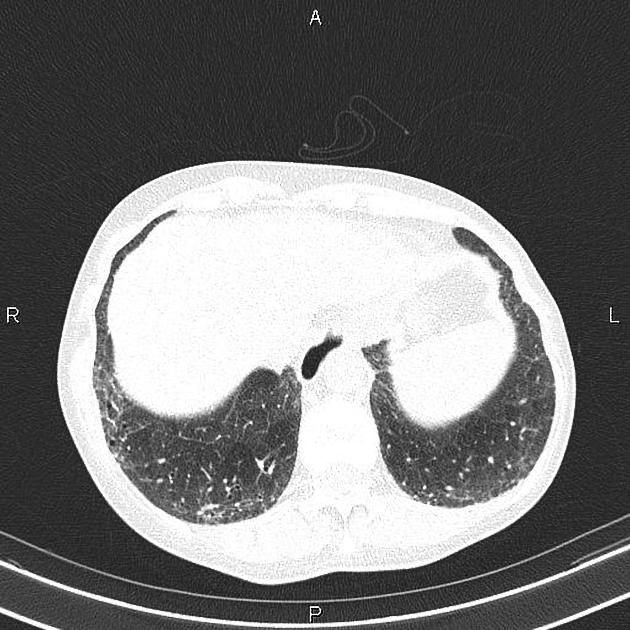
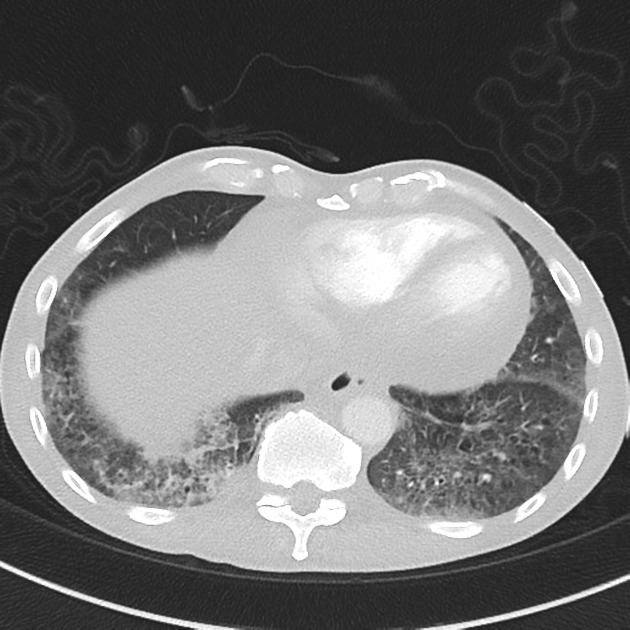
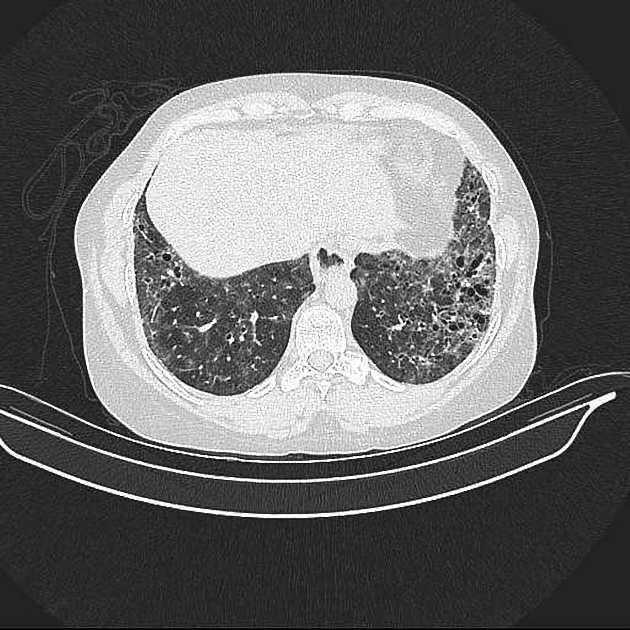
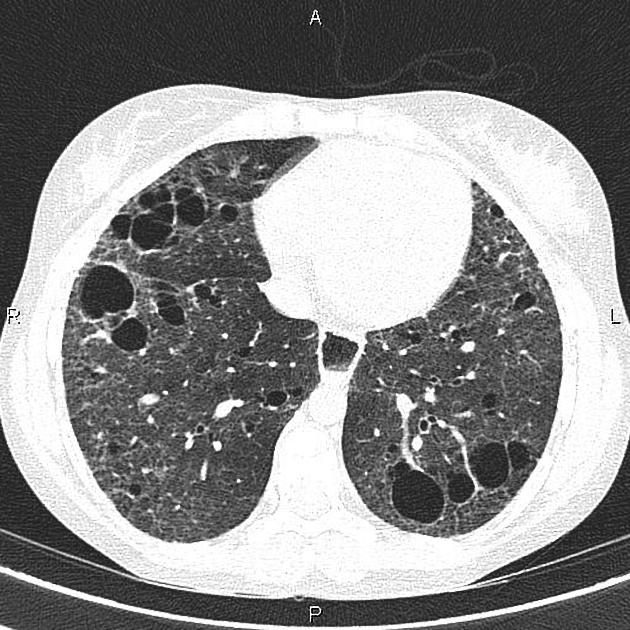
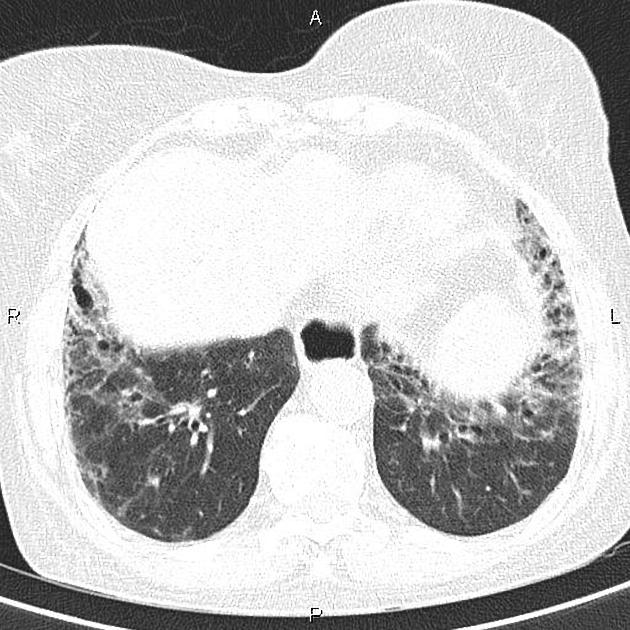
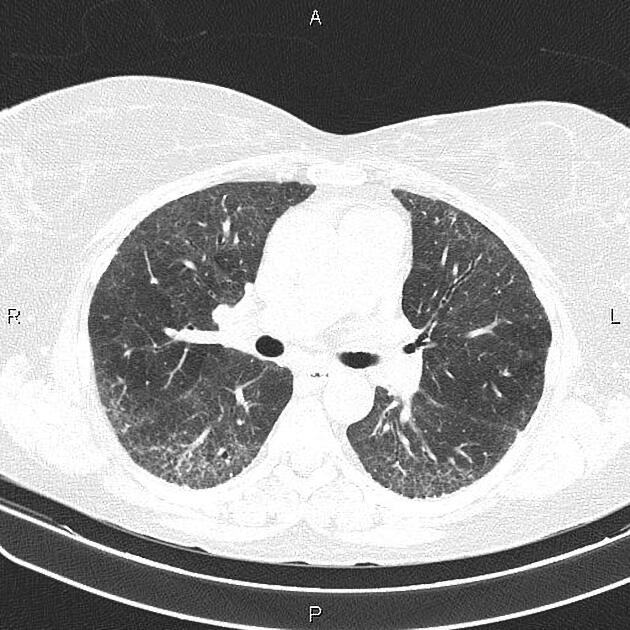
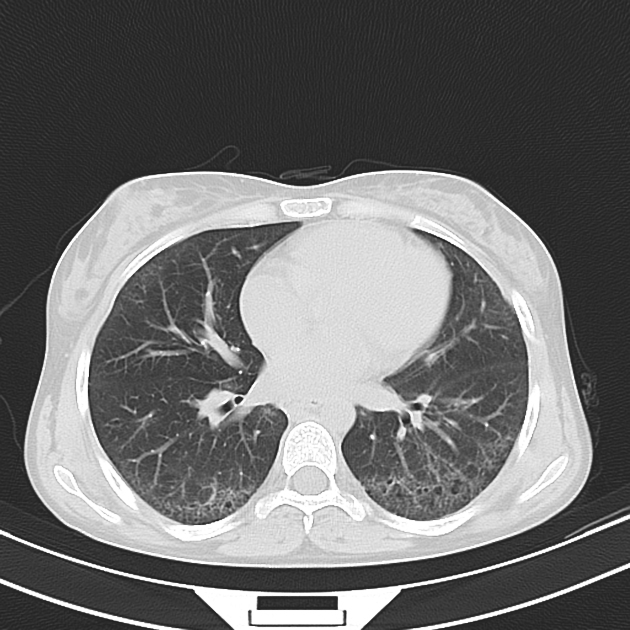
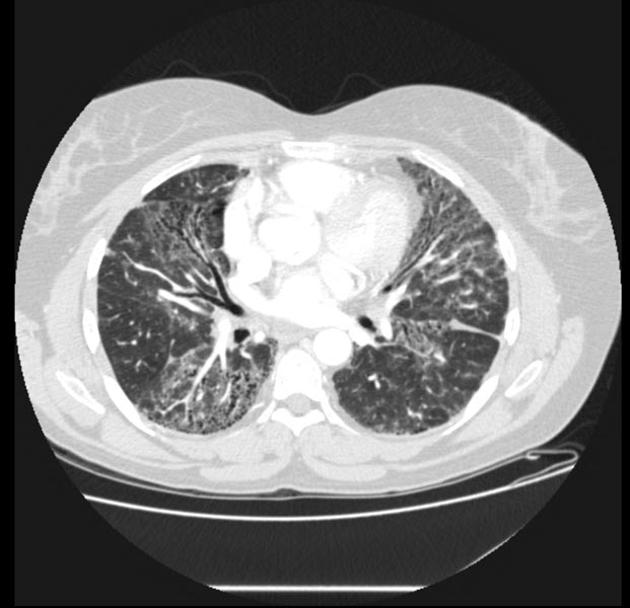
On HRCT, scleroderma may appear in either a non-specific interstitial pneumonitis (NSIP) (more common 5) or usual interstitial pneumonitis (UIP) pattern. Interstitial lung changes in scleroderma are less extensive and less coarse than those with idiopathic pulmonary fibrosis 3 with most patients only show a limited portion of lung involvement (less than 10% in half of cases) 1.
early stages may show ground glass changes
later stages may show honeycombing and evidence of lung volume loss
lung bases and sub-pleural regions typically involved 4
cysts may be present measuring 1-5 cm in diameter 4
pleural effusions are usually not a feature
CT may also show features of pulmonary hypertension and mediastinal lymphadenopathy 10,11
four corners sign: bilateral anterolateral upper lobe and posterosuperior lower lobe inflammation and fibrosis has been reported as a specific finding to help differentiate scleroderma from idiopathic pulmonary fibrosis 12
-
tends to be cylindrical in general
maybe present in around 60% of patients according to one study 8
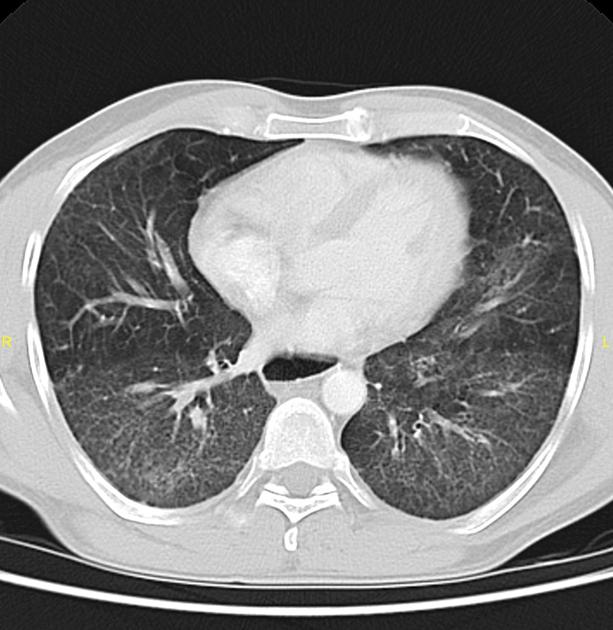
a dilated esophagus is not an uncommon finding
Treatment and prognosis
Pulmonary involvement is incurable but some compounds may improve or halt progression. These include cyclophosphamide, glucocorticoids or N-acetylcysteine, although the data is inconclusive 5.
Complications
pulmonary arterial hypertension is seen in up to 50% of patients and may develop without evidence of interstitial fibrosis 2,4
higher incidence of lung cancer, particularly adenocarcinoma 4
rupture of pulmonary cysts resulting in a pneumothorax 4
The cause of death is often from aspiration pneumonia.
Differential diagnosis
On HRCT, the differential is essentially that of usual interstitial pneumonia and lower zone pulmonary fibrosis.
A helpful clue to the diagnosis on CT is the presence of a dilated esophagus.
Pleural effusions are distinctly uncommon in scleroderma and would favor the diagnosis of either rheumatoid lung or SLE 4.


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.