
Idiopathic pulmonary fibrosis (IPF) is a clinical syndrome and considered the most common and the most lethal form of pulmonary fibrosis corresponding to the histologic and imaging pattern of usual interstitial pneumonia. It is more common in elderly men and diagnosed by:
histological or imaging pattern of usual interstitial pneumonia (UIP) and
absence of alternative causes such as drug toxicity, environmental exposure (e.g. asbestos) or collagen vascular disease (e.g. scleroderma, rheumatoid arthritis)
On this page:
Epidemiology
It tends to be commoner in males, with most cases presenting in those over 60 years of age. Though, it might be also seen in middle aged adults, particularly in those with familial risk for pulmonary fibrosis 15.
Clinical presentation
Patients typically present with progressive dyspnea on exertion and chronic dry cough, usually over a period of 24 months before diagnosis. Other associated features might include chest pain, fatigue, malaise, and weight loss.
Physical examination usually reveals fine end-inspiratory crackles and in severe cases finger clubbing.
Pulmonary function test results may be normal in mild disease or show restriction pattern (i.e. reduced vital capacity and total lung capacity but near normal residual volume). Lung function tests show a restrictive pattern with a decreased diffusing capacity of the lungs for carbon monoxide (DLCO).
Diagnostic criteria
A multidisciplinary approach in a tertiary setting is strongly advised. Contributions from pulmonologists, chest radiologists, and chest pathologists are crucial in reaching the correct diagnosis of IPF.
In 2000, the American Thoracic Society (ATS) and European Respiratory Society (ERS) jointly agreed major and minor criteria for the diagnosis of IPF in the absence of a surgical lung biopsy 5:
Major criteria
exclusion of other known causes of interstitial lung disease (e.g. toxic effects of certain drugs, environmental exposures, connective tissue diseases)
abnormal results of pulmonary function studies, including evidence of restriction (reduced vital capacity, often with an increased FEV1/FVC ratio) and impaired gas exchange (increased PaO2, decreased PaO2 with rest or exercise, or decreased DLCO)
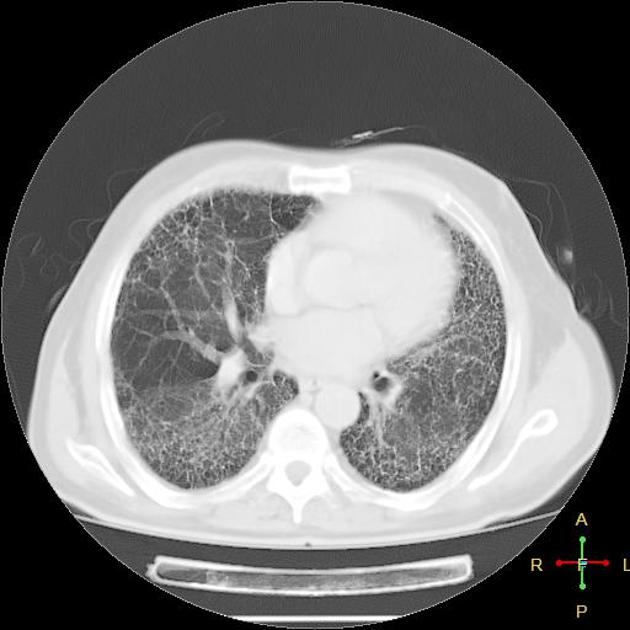
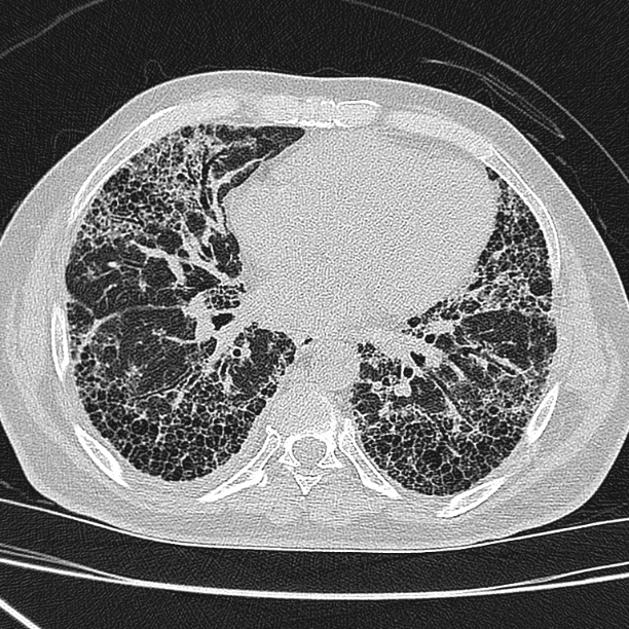
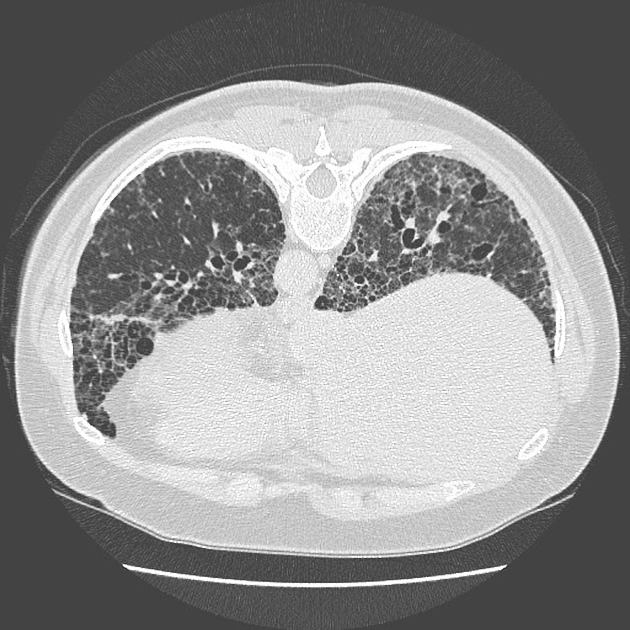
bibasilar reticular abnormalities with minimal ground-glass opacities at high-resolution CT: definite UIP pattern on HRCT chest
transbronchial lung biopsy or bronchoalveolar lavage shows no features to support an alternative diagnosis
Minor criteria
age >50 years
insidious onset of otherwise unexplained dyspnea on exertion
duration of illness >3 months
bibasilar inspiratory crackles (dry or “Velcro” type)
In 2018, these criteria were revised in a collaborative statement by the ATS, ERS, Japanese Respiratory Society (JRS) and Latin American Thoracic Association (ALAT) 12. The major and minor criteria were eliminated and only the following diagnostic criteria remain:
Exclusion of other known causes of interstitial lung disease (e.g. domestic and occupational environmental exposures, connective tissue disease, and drug toxicity).
The presence of a UIP pattern on HRCT in patients not subjected to surgical lung biopsy (diagnostic categories of UIP pattern based on HRCT chest - Fleischner Society guideline 2018).
Specific combinations of HRCT and surgical lung biopsy pattern in patients subjected to surgical lung biopsy.
Pathology
IPF, as the name states, is idiopathic, however, there is an association with concurrent or previous history of smoking in 60% of patients and genetic factors. Up to 5–20% of patients with IPF have a family history of interstitial lung disease (ILD) or pulmonary fibrosis. It has been shown that fibroblasts in this process demonstrate neoplastic or neoproliferative characteristics 11.
Genetics
The rs35705950 single-nucleotide polymorphism (SNP)—a promoter site of an airway mucin gene (MUC5B)—is strongly associated with IPF and familial pulmonary fibrosis and not seen in other secondary causes of lung fibrosis. Positive rs35705950 SNP in IPF patients is associated with slightly better prognosis and outcome.
Histopathology
Histology shows a UIP pattern which is characterized by spatial and temporal heterogeneity. One of the hallmarks is the absence of inflammation. Spatial heterogeneity denotes biopsy sample showing patchy lung involvement with normal lung interspace between diseased lung. Temporal heterogeneity denotes different stages of disease seen on a single specimen, including normal lung, interstitial fibrosis and fibroblastic foci 4.
Radiographic features
The CT imaging findings complement the histology. It is more correct to describe the characteristic imaging pattern as UIP rather than IPF, the latter term assigned for the idiopathic clinical syndrome of UIP.
A UIP-pattern of fibrosis is characterized by honeycombing cysts and reticular septal thickening with subpleural and posterior basal predominance. Traction bronchiectasis can also be observed, however, this is a general feature of fibrosis not specific to the UIP-pattern 4. In a subgroup of patients, the imaging findings of UIP overlap with NSIP and biopsy may be necessary to obtain the correct diagnosis.
Treatment and prognosis
The clinical course is that of gradual deterioration and the condition carries a rather poor prognosis with median survival ranging from 2.5 to 3.5 years from the time of diagnosis (at the time of initial writing) 2. Some reports have suggested a slowing of progression with treatment by pirfenidone or nintedanib 7,8.
Differential diagnosis
Consider pulmonary fibrosis due to a known cause, such as:
collagen vascular disease-related usual interstitial pneumonia (UIP)
non-specific interstitial pneumonia pattern (especially fibrotic non-specific interstitial pneumonia)
-
fibrotic hypersensitivity pneumonitis
hypersensitivity pneumonitis usually involves the mid and upper zones of the lung, and also the presence of centrilobular nodules and areas of air trapping are very useful hints to differentiate it from UIP
UIP cases are also thought to have honeycombing and peripheral or lower lung zone predominance of disease, and less likely to have micronodules
amiodarone lung fibrosis: helpful clues are the presence of hyperdense pulmonary nodules or hyperdense liver on a non-contrast CT
systemic sclerosis: presence of patulous esophagus and correlation with hand radiographs if available can be helpful
asbestosis: bilateral pleural plaques with or without calcification or peritoneal calcification are helpful in diagnosis
combined pulmonary fibrosis and emphysema (CPFE): especially if there is added upper lobe-predominant emphysema
congenital, e.g. Hermansky-Pudlak syndrome
Practical points
Classification tends to ignore the extent and variety of lung injury in IPF; e.g. damage to alveolar lining cells, pulmonary vessels, and pleura may also be present. There is a marked decrease in the volume of alveolar gas and damage and loss of small airways, associated with mosaic attenuation which may mislead radiologists into suggesting an alternative diagnosis 19.
See also
cryptogenic fibrosing alveolitis: some authors consider this term to be synonymous with IPF 6



 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.