
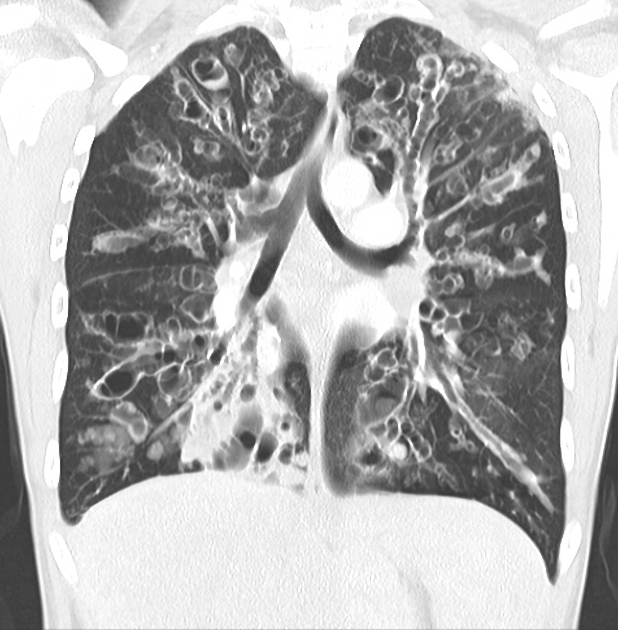
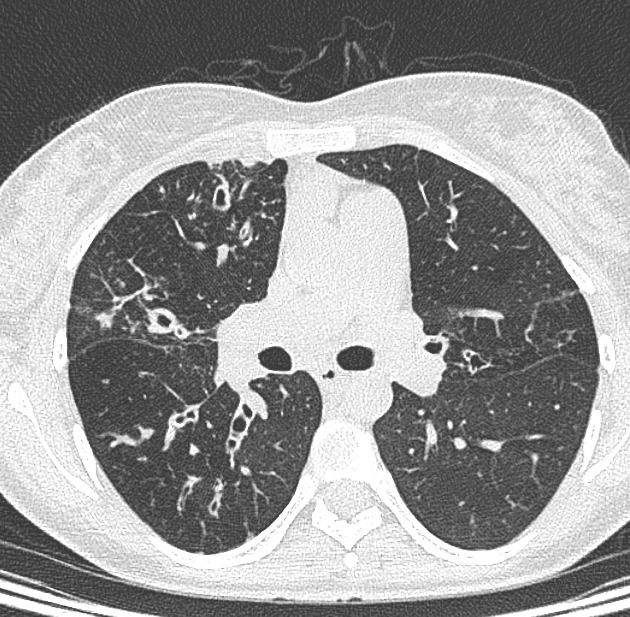
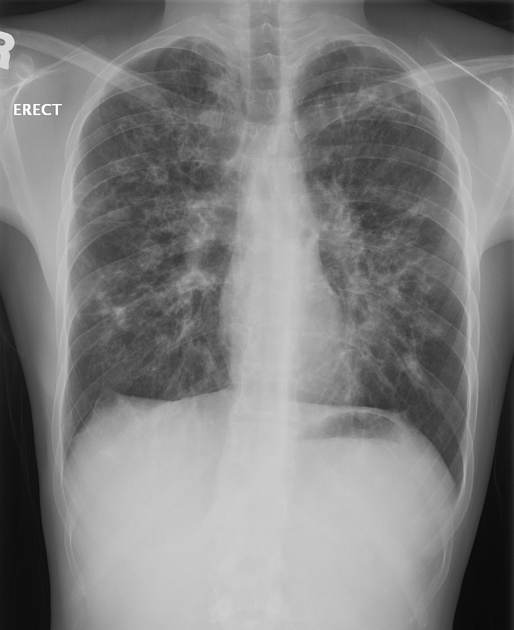
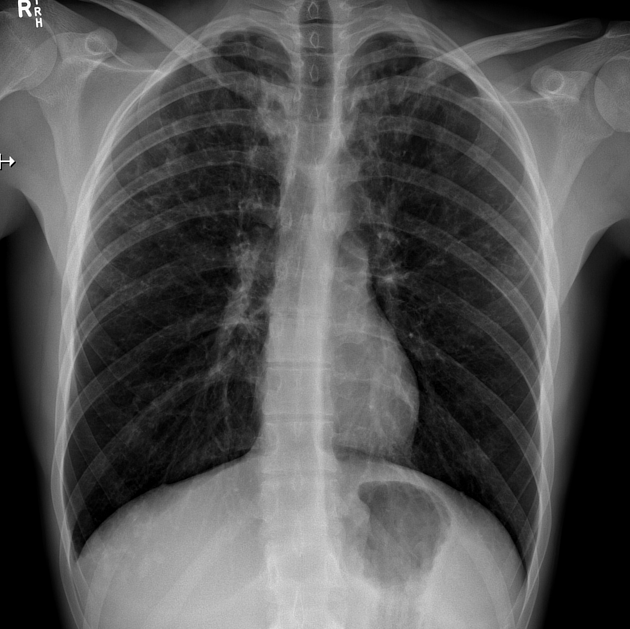
Cystic fibrosis (CF), also called mucoviscidosis, is an autosomal recessive genetic disease that affects the exocrine function of the lungs, liver, pancreas, small bowel, sweat glands, and urogenital system.
This article is a general discussion of the disease. Each organ system are discussed separately, see:
-
recurrent bacterial infection
-
-
pancreas (most common abdominal organ involved 9)
exocrine and endocrine insufficiency (type 3c diabetes mellitus)
pancreatitis (acute and chronic)
-
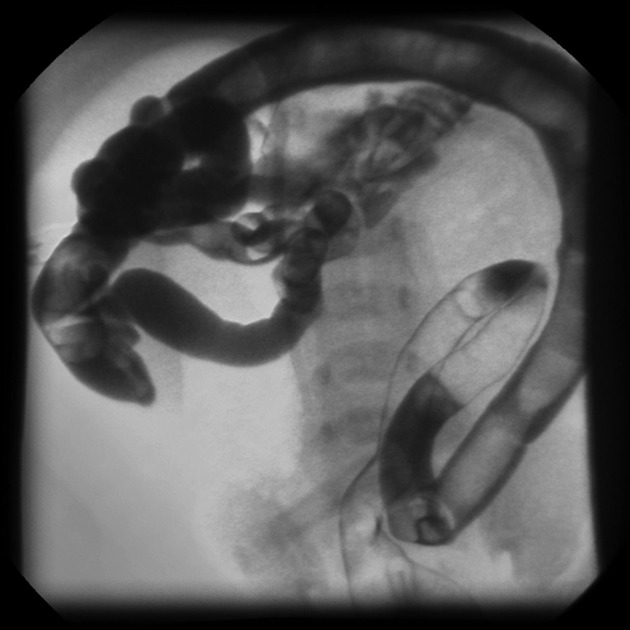
liver
focal biliary and multilobular cirrhosis
-
biliary system
-
gastrointestinal tract
meconium ileus: 10-20%
esophageal dysfunction and gastro-esophageal reflux
distension of appendix but reduced risk of appendicitis
-
-
urogenital tract manifestations 11
bilateral seminal vesicle agenesis
hypoplasia or agenesis of the ductus deferens
hypoplasia or agenesis of the tail and body of the epididymis
On this page:
Epidemiology
Cystic fibrosis is the most common genetic disease affecting European population with an incidence of approximately 1:2000-3500 live births 5.
Clinical presentation
The diagnosis may be suspected antenatally due to the presence of echogenic bowel on antenatal ultrasound, or due to genetic testing of the parents.
In many countries, the presence of cystic fibrosis is tested for immediately after birth with a sweat test (positive sweat chloride test Cl >60 mEq/L) 5. Alternatively, genetic testing is also available.
The diagnosis usually becomes evident in infancy, with presentations including 12:
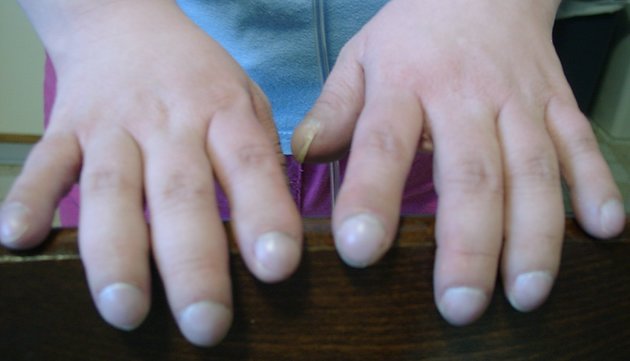
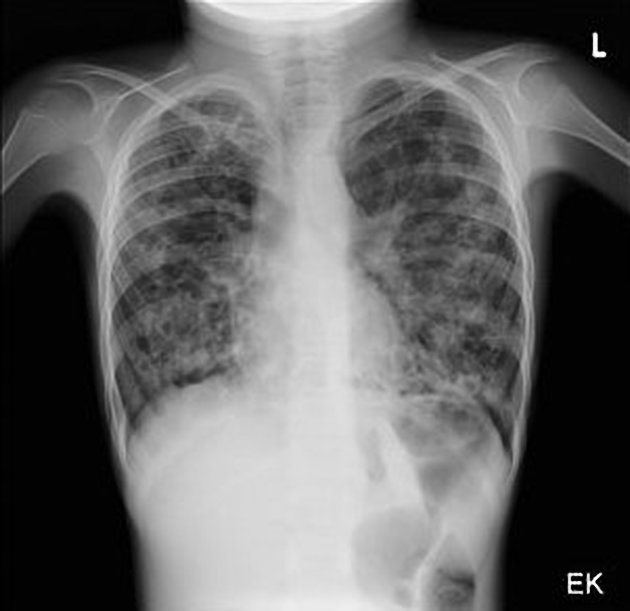
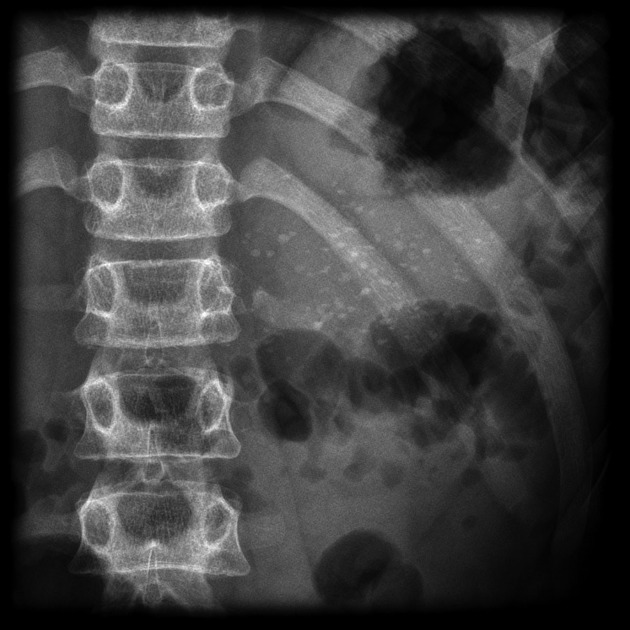
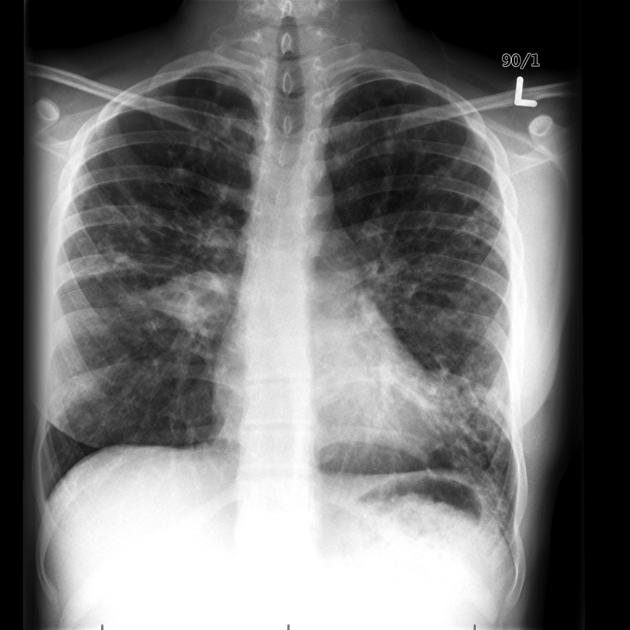
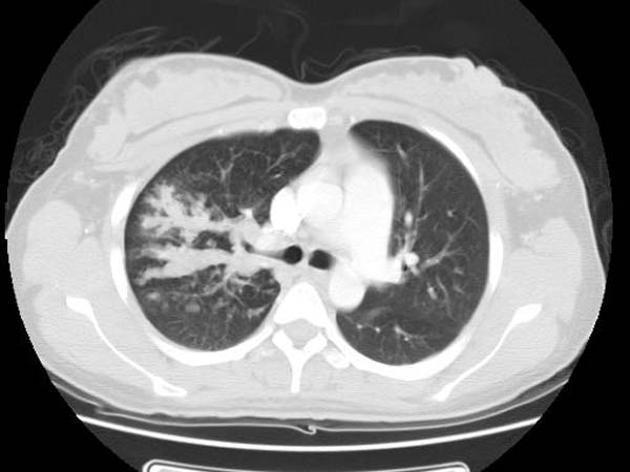
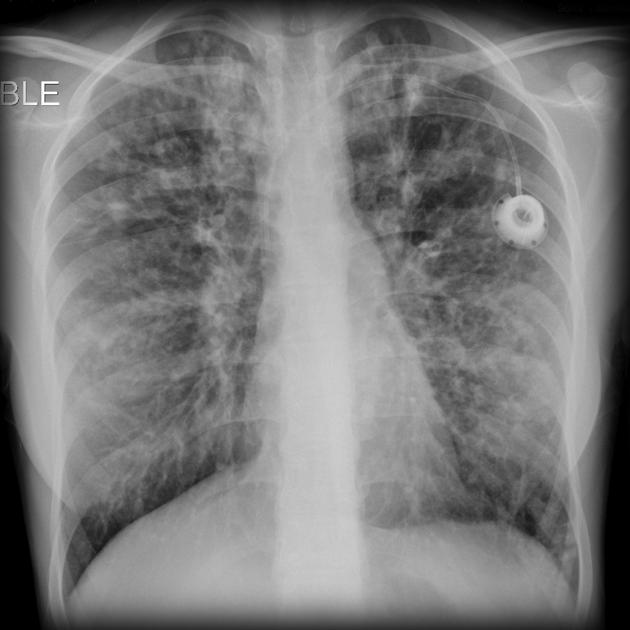
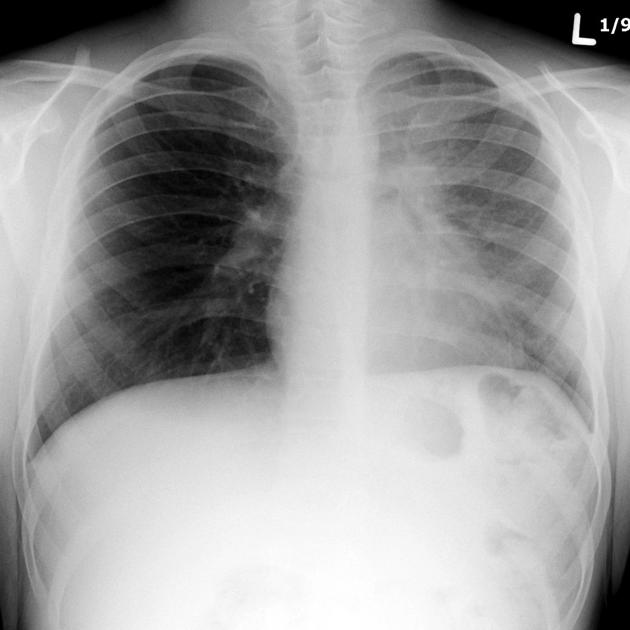
recurrent pulmonary infection
steatorrhea
poor weight gain
Pathology
Genetics
Cystic fibrosis is an autosomal recessive inherited genetic disorder that results from a homozygous defect of the cystic fibrosis transmembrane regulator (CFTR) gene on chromosome 7q31.2. The gene encodes for the corresponding CFTR protein, which regulates chloride ion transport across cell membranes. There are over 2000 mutations of the CTFR gene identified to date, with the most common being delF508 (deletion of the codon for phenylalanine at the 508 position) affecting 66% of cases 6,14.
CFTR gene mutations are classified into 6 categories based on the synthesized protein structure and function as follows 14,15,17:
class I: mutations resulting in premature stop codons; CFTR expression is severely reduced or absent
class II: mutations resulting in CFTR misfolding and increased degradation; functional CFTR reacting the cell surface is reduced
class III: mutations that impair regulation of the CFTR channel; abnormal gating with reduced opening
class IV: mutations that impair CFTR ion conductance
class V: mutations of CFTR promotor or splicing; CFTR protein is normal in structure and function, but reduced in number
class VI: mutations that reduce stability of CFTR; increased turnover of CFTR and reduced duration at the cell surface
The precise way in which CFTR mutations cause disease is complex and still under investigation. However, the most widely accepted explanation is that these mutations reduce extracellular chloride ion transport, leading to production of abnormally thickened mucus and eventual organ dysfunction 14,15. This is now known to be an oversimplification, as it has been shown that the CFTR protein also regulates the transport of sodium, bicarbonate, and glutathione 15.
Unlike in other tissues, CFTR in sweat glands function in the opposite way, responsible for intracellular chloride ion transport 16. In patients with cystic fibrosis, the CFTR mutation results in excess chloride, sodium and fluid loss onto the skin surface, hence the use of the sweat test for diagnosis 16.
Treatment and prognosis
Early treatment is essential and responsible for the dramatic increase in life expectancy, now reaching 40 years or more.
Treatment options include 7,10,13:
dietary changes, including pancreatic enzyme and vitamin supplementation
physiotherapy and airway clearance techniques
-
cystic fibrosis transmembrane regulator (CFTR) modulators
-
for delF508 mutation:
ivacaftor and lumacaftor combination therapy
ivacaftor and tezacaftor combination therapy
elecacaftor-tezacaftor-ivacaftor combination therapy
for G551D mutation: ivacaftor monotherapy
-
anti-inflammatory therapy (e.g. azithromycin)
antibiotics, often multiple agents administered for prolonged courses
oral and inhaled corticosteroids
lung transplantation
specific management of complications (e.g. diabetes mellitus, hemoptysis, distal intestinal obstruction syndrome (DIOS), etc.)
Both transplanted and non-transplanted patients with cystic fibrosis are at increased risk of some malignancies 8:
gastrointestinal malignancy, in particular esophageal, gastric, small bowel, and colorectal cancer
gallbladder and extrahepatic biliary tree malignancies





 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.