
Leigh syndrome, also known as subacute necrotizing encephalomyelopathy (SNEM), is a mitochondrial disorder characterized by progressive neurodegeneration, mitochondrial dysfunction, and bilateral central nervous system lesions, that invariably leads to death, usually in childhood.
On this page:
Epidemiology
Leigh syndrome is rare, encountered in approximately 1 in 40,000 births, although some populations have much higher incidence (e.g. in Quebec in Canada) 9. There is no known gender or racial predilection 9.
Clinical presentation
Typically, symptoms become evident before the age of 2 years, with a presentation in later childhood (juvenile form) or adulthood (adult form) being very rare 11. Later onset forms tend to progress more slowly 11.
The clinical presentation is incredibly heterogenous and often triggered by a metabolic challenge (e.g. infection), but potential features include (in approximate order of most to least common) 6,9-13,18:
-
psychomotor or developmental delay/regression
overall the most common clinical feature
more common in early onset disease
hypotonia
respiratory dysfunction
poor feeding and dysphagia
seizures (focal and/or generalized types) and epilepsy
-
neuromuscular weakness
more common in later onset disease
ocular signs: nystagmus, ptosis, ophthalmoplegia, optic atrophy, retinopathy
-
ataxia
more common in later onset disease
dystonia
sensorineural hearing loss
Less commonly, there is involvement of organs beyond the central nervous system, including cardiac, gastrointestinal and renal manifestations 11,13. Hypertrichosis has been described in some patients specifically with a SURF1 mutation 18.
Pathology
Genetics
Leigh syndrome is one of many mitochondrial disorders. It is caused by dysfunction or deficiency of one or multiple of the complexes comprising the electron transport chain in mitochondria 11.
These complexes are encoded by either nuclear DNA (nDNA) or mitochondrial DNA (mtDNA), and thus, mutations in various genes from each genome can result in Leigh syndrome 8-11,13. Later onset phenotypes are more often associated with mtDNA mutations 18.
Commonly implicated genes in Leigh syndrome include 1,10,11,13:
-
mtDNA:
MT-ATP6 gene which encodes for complex V
MT-ND genes (e.g. MT-ND5) which encode for complex I
nDNA: SURF1 gene which encodes for complex IV
Given the wide range of genes, there is a range of inheritance patterns observed in Leigh syndrome 9,11. This includes mitochondrial (i.e. maternal) or autosomal recessive patterns of inheritance, and very rarely, also X-linked recessive and autosomal dominant patterns of inheritance 9,11.
Microscopic appearance
Brain biopsy is rarely performed but shows features of chronic energy deprivation leading to histological features such as 3:
spongiform degeneration
capillary proliferation
neuronal loss
These findings are similar to those seen in infarction 4.
Markers
CSF and serum lactate levels are usually elevated 10,13.
Radiographic features
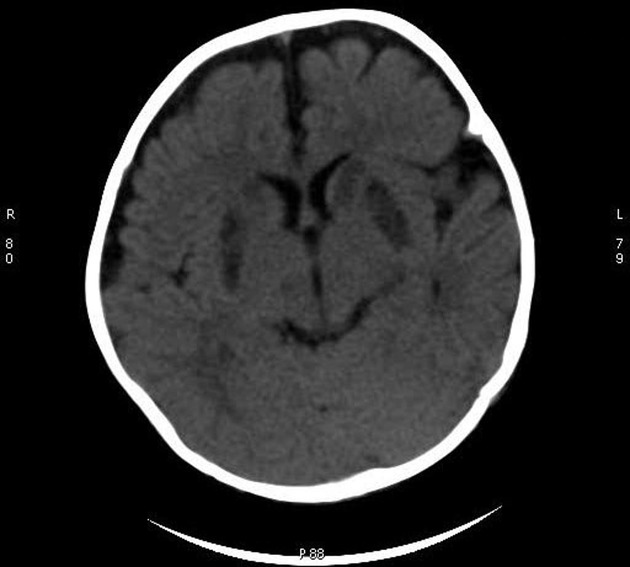
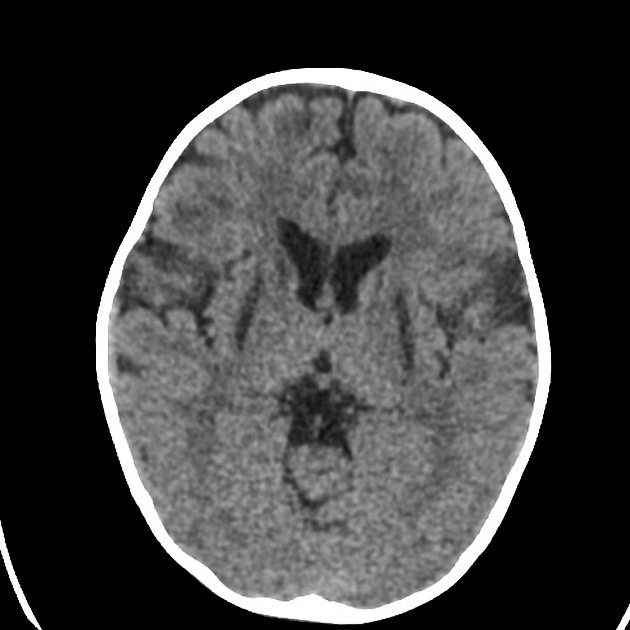
CT
CT demonstrates regions of low-density matching areas of the abnormal T2 signal on MRI (see below) 5. Occasionally some of these areas can show contrast enhancement 5.
MRI
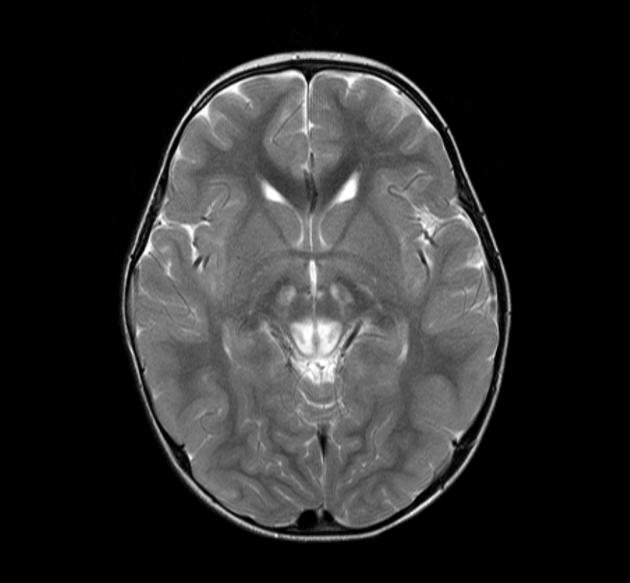
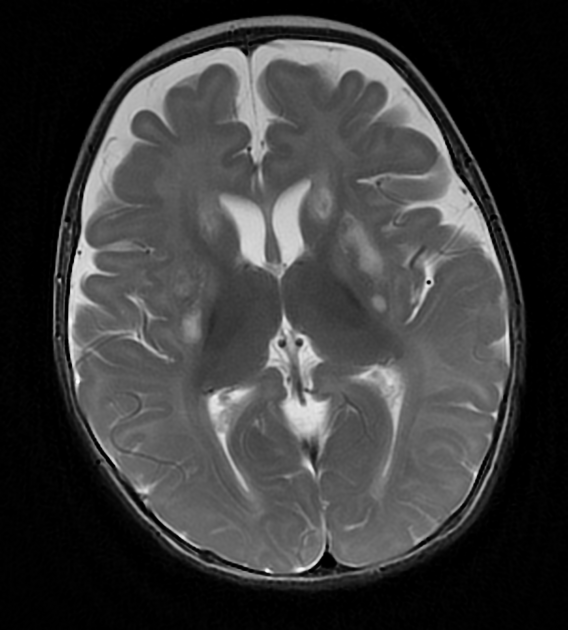
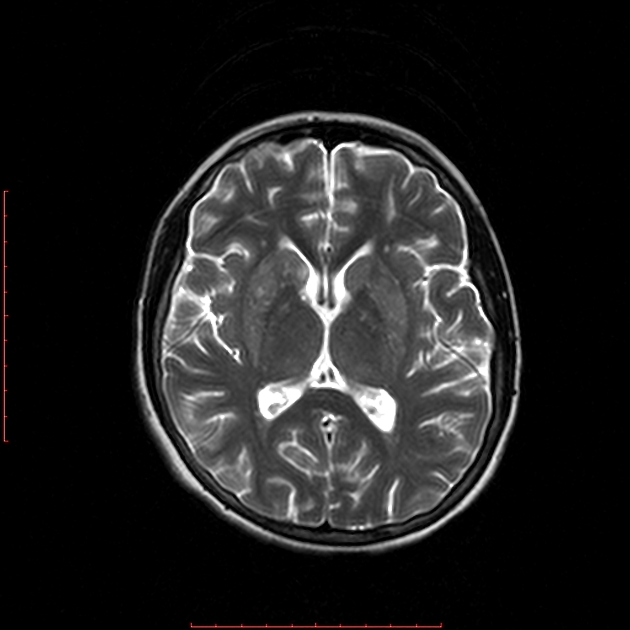
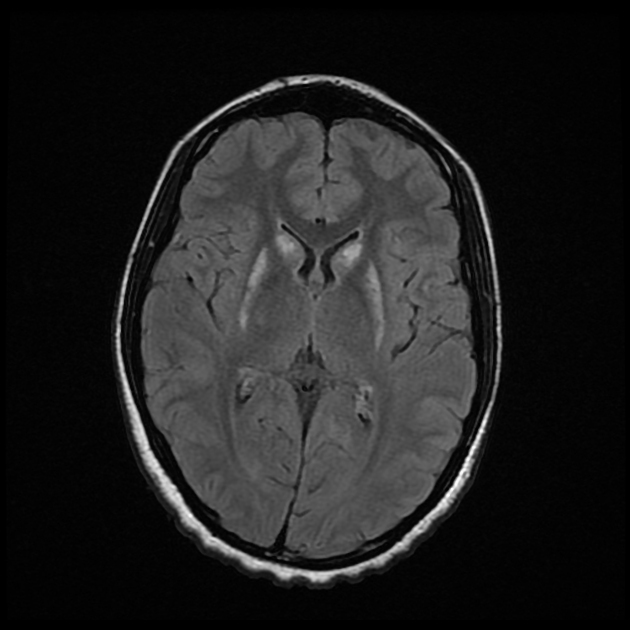
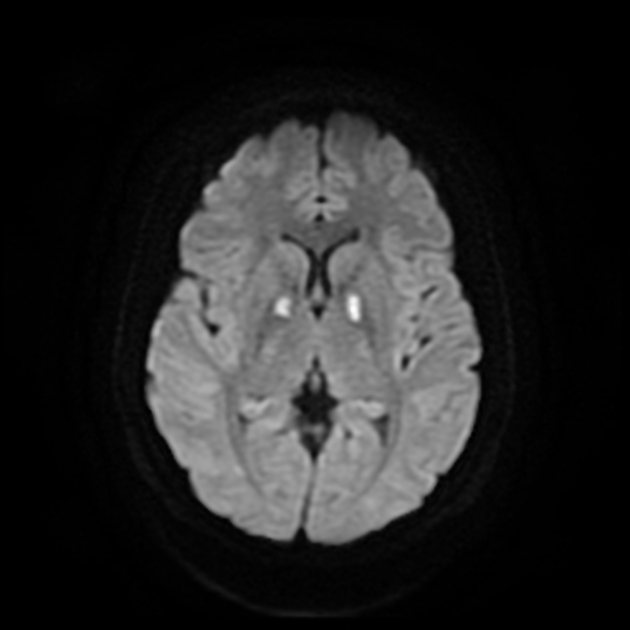
MRI abnormalities are heterogeneous, although no definite differences have been consistently noted between different mutations 8,13. Generally, the distribution tends to be symmetrical and some lesions may regress with time, while others may be permanent and evolve 13,16.
-
T2: high signal 3,10,11,13,18
basal ganglia (most common), in particular the putamen
-
brainstem, can affect any level but in particular the midbrain (e.g. periaqueductal grey matter) and medulla oblongata
the double panda sign has been rarely reported 17
subthalamic nuclei and dentate nuclei 19
involvement of cerebral white matter, cerebellar white matter, or spinal cord is uncommon 19
T1: usually demonstrates reduced signal in regions of T2 high signal, although some areas of hyperintensity can be seen, as can some enhancement post-gadolinium administration
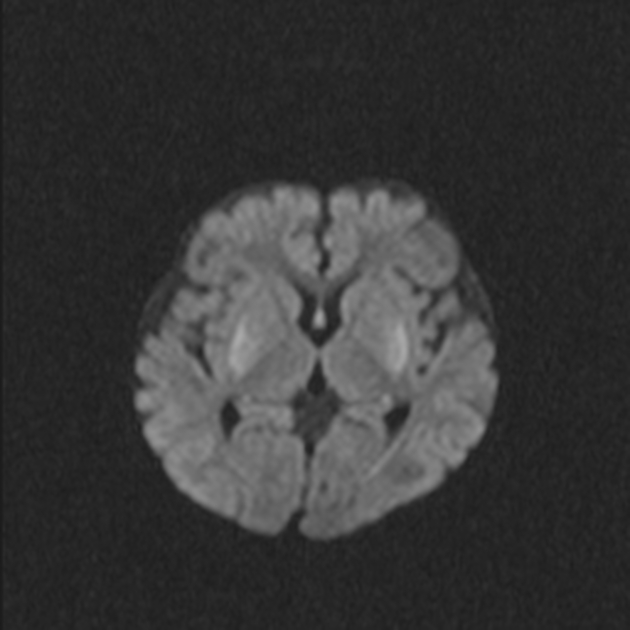
DWI: in the acute setting high diffusion signal may be evident (so-called 'stroke-like' lesions)
-
MR spectroscopy
Treatment and prognosis
There is no disease-modifying therapy available 10,18, although a 'mitochondrial cocktail' is often prescribed, variably consisting of components such as thiamine (vitamin B1), coenzyme Q-10, riboflavin (vitamin B2) and vitamin C 14,15,18. Generally, medications which are mitochondrial toxins (e.g. sodium valproate) should be avoided 15.
Prognosis is poor, with death usually occurring in childhood. The later the onset, the slower the deterioration. Death is most frequently due to respiratory failure 6.
The factors associated with a worse outcome are 10,13:
disease onset before 6 months of age
admission to an intensive care
brainstem lesions
MRS lactate peak
History and etymology
It is named after Archibald Denis Leigh (1915-1998), a British psychiatrist and neuropathologist, who first described the condition in 1951 2,9,18.
Differential diagnosis
-
similar appearance but typically presents in a very different patient demographic
mammillary bodies not typically involved in Leigh syndrome
enhancement more common in Wernicke encephalopathy
hemorrhagic change more common in Wernicke encephalopathy 4
-
other mitochondrial disorders (e.g. MEGDEL syndrome)
brainstem and basal ganglia involvement less pronounced
-
acute necrotizing encephalitis of childhood
lactate levels are usually normal
toxic encephalopathies (e.g. carbon monoxide poisoning)




 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.