
Congenital pulmonary airway malformations (CPAM) are multicystic masses of segmental lung tissue with abnormal bronchial proliferation. CPAMs are considered part of the spectrum of bronchopulmonary foregut malformations.
On this page:
Terminology
CPAMs were previously termed congenital cystic adenomatoid malformations (CCAM).
Epidemiology
They account for ~25% of congenital lung lesions. The estimated incidence is approximately 1:1500-4000 live births and there is a male predominance.
Associations
hybrid lesion: i.e. CPAM and pulmonary sequestration
-
~10% of pediatric lung cancers have a history of CPAM 13
mucinous adenocarcinoma associated with type 1 CPAM 14
pleuropulmonary blastoma associated with type 4 CPAM 14
Clinical presentation
The diagnosis is usually either made on antenatal ultrasound, or in the neonatal period on the investigation of progressive respiratory distress 3,4. If large, they may cause pulmonary hypoplasia, with a resultant poor prognosis.
In cases where the abnormality is small, the diagnosis may not be made for many years or even until adulthood. When it does become apparent, it is usually as a result of recurrent chest infection 3,4.
Pathology
The condition results from failure of normal bronchoalveolar development with a hamartomatous proliferation of terminal respiratory units in a gland-like pattern (adenomatoid) without proper alveolar formation.
These lesions have intracystic communications and, unlike bronchogenic cysts, can also have a connection to the tracheobronchial tree.
Location
Lesions are usually unilateral and involve a single lobe. Although there is no well-documented lobar predilection, they appear less frequently in the middle lobe 3.
Classification
According to the Stocker classification, there are five subtypes based on cyst size 15:
-
type 0
very rare, lethal postnatally
acinar dysgenesis or dysplasia 11
represents global arrest of lung development 12
-
type I
most common: 70% of cases 3
large cysts, lined by pseudo-stratified columnar epithelium 15
one or more dominant cysts: 2-10 cm in size
may be surrounded by smaller cysts
-
type II
15-20% of cases 3
cysts are <2 cm in diameter
-
associated with other abnormalities
-
type III
~10% of cases
microcysts: <5 mm in diameter
typically involves an entire lobe
has a poorer prognosis
-
type IV
large cysts (typically >10 cm) lined by flattened epithelium (type 1 and 2 pneumocytes) 15
typically affects a single lobe
indistinguishable from type I on imaging 11
Microscopic appearance
Histologically, they are characterized by adenomatoid proliferation of bronchiole-like structures and macro- or microcysts lined by columnar or cuboidal epithelium and absence of cartilage and bronchial glands.
Radiographic features
The appearance of CPAM will vary depending on the type.
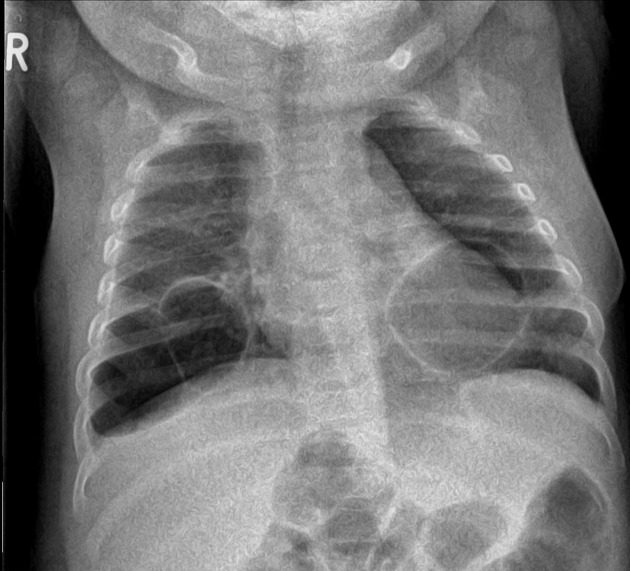
Plain radiograph
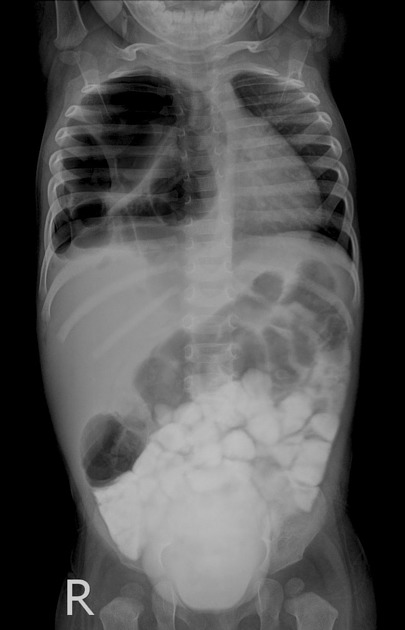
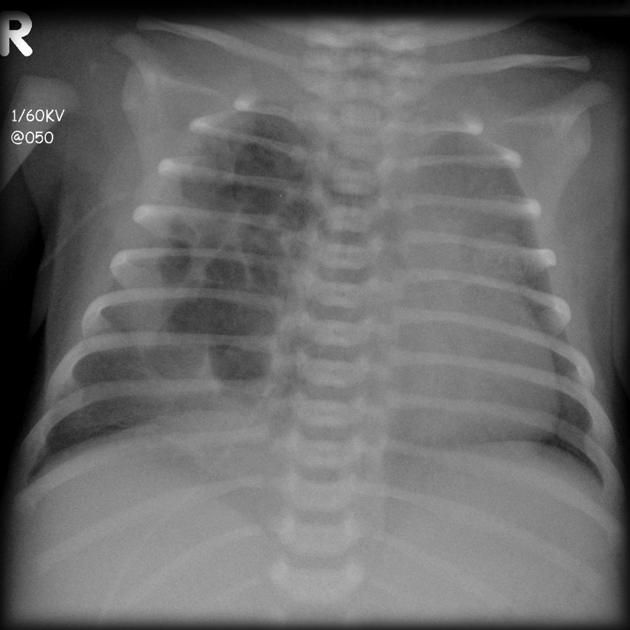
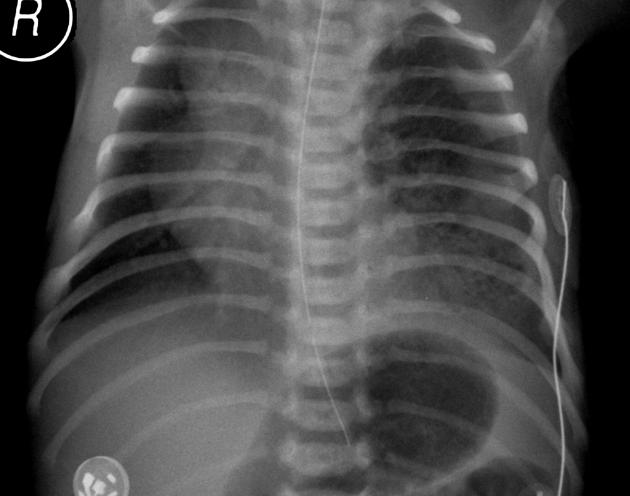
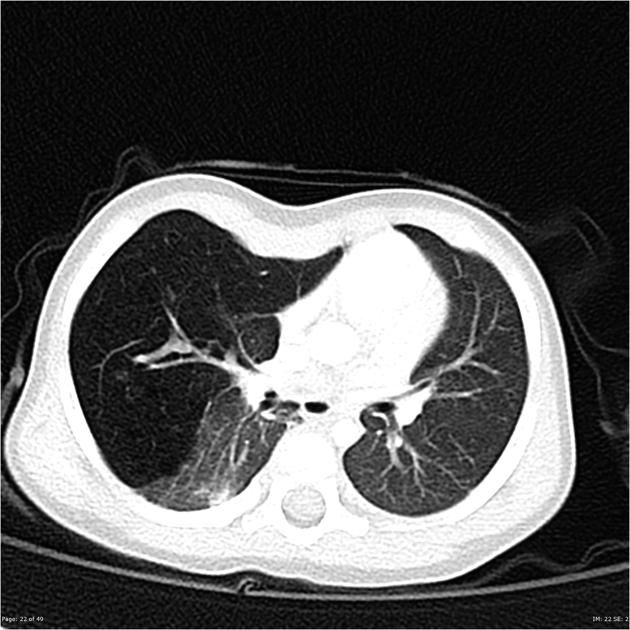
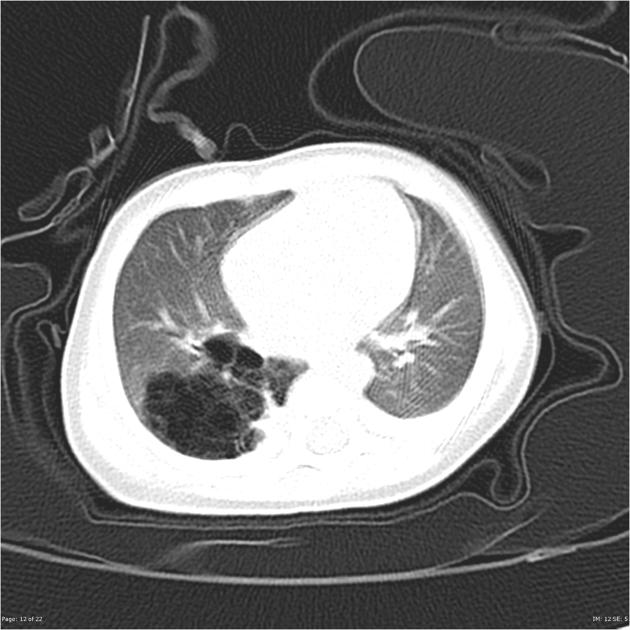
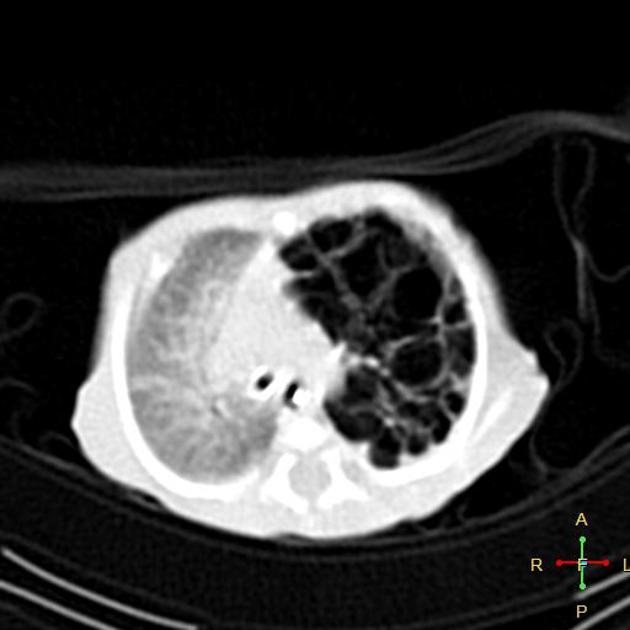
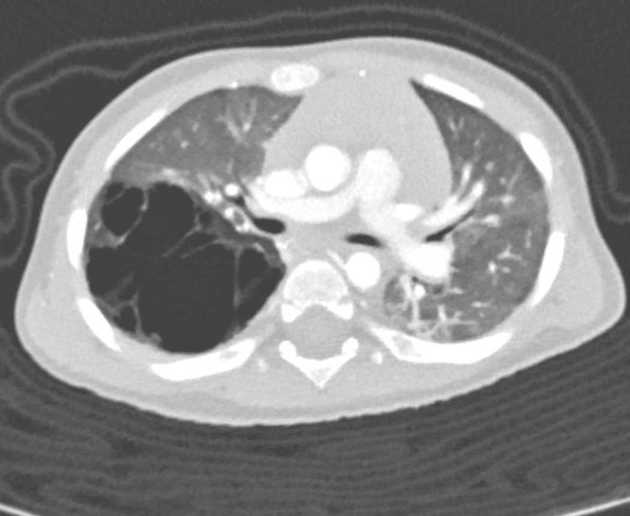
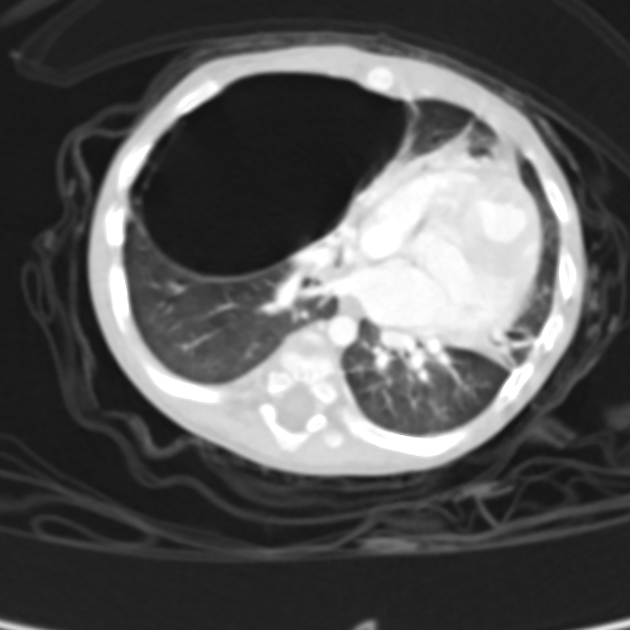
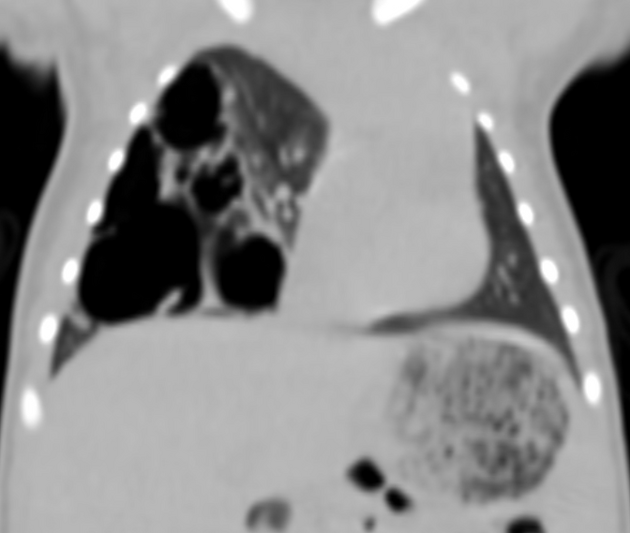
Chest radiographs in type I and II CPAM may demonstrate a multicystic (air-filled) lesion. Large lesions may cause a mass effect with resultant mediastinal shift, depression, and even inversion of the diaphragm. In the early neonatal period, the cysts may be completely or partially fluid-filled, in which case the lesion may appear solid or with air-fluid levels. Lesions may change in size on interval imaging (expand from collateral ventilation via pores of Kohn). Type III lesions appear solid.
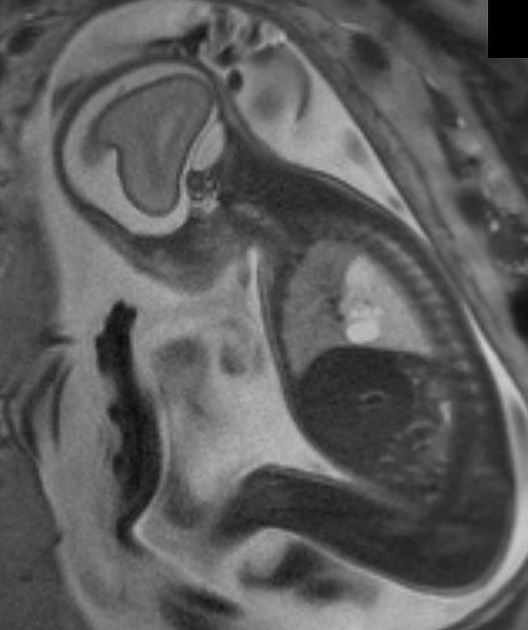
Ultrasound
CPAM appears as an isolated cystic or solid intrathoracic mass. A solid thoracic mass is usually indicative of a type III CPAM and is typically hyperechoic. There can be a mass effect where the heart may appear displaced to the opposite side. Alternatively, the lesion may remain stable in size, or even regress 5.
Hydrops fetalis and polyhydramnios may develop and may be detected on antenatal ultrasound as ancillary sonographic features 3.
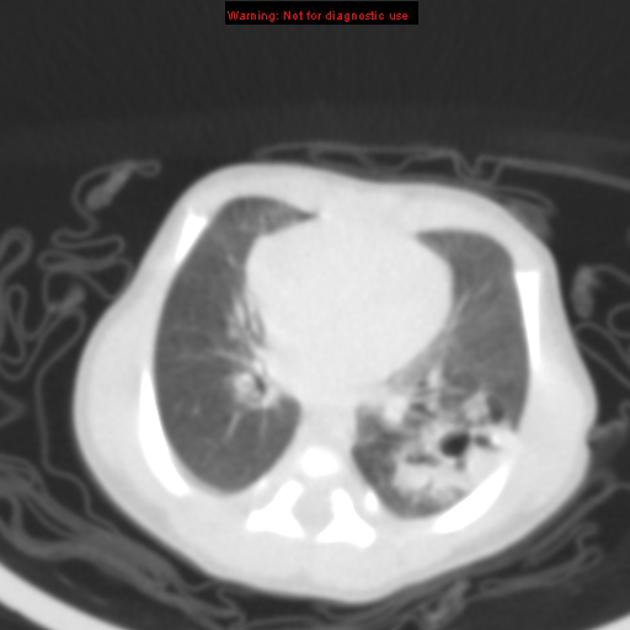
CT
CT has a number of roles in the management of CPAM. First, it more accurately delineates the location and extent of the lesion. Secondly, and most importantly in surgical candidates, CT angiography is able to identify systemic arterial supply if present.
Appearance reflects the underlying type, and a type III lesion can appear as a consolidation.
Treatment and prognosis
There is a wide spectrum of prognosis.
Surgery (elective lobectomy) is the treatment of choice in symptomatic patients, both in those presenting early with respiratory compromise and in those presenting later with recurrent infections 3. Type I lesions have the best prognosis.
In the setting of a small stable asymptomatic lesion, surgical excision is more controversial. Advocates for excision quote the reported risk of developing malignancies within the lesion (see above). An alternative approach is to watch and wait. There are reports of spontaneous regression, particularly in those serially followed up on antenatal ultrasound 7,10.
Complications
Potential postnatal complications include:
recurrent pneumothorax
-
possible incidence of certain malignancies, which include 3:
Potential in utero complications include:
hydrops fetalis may rarely develop when there is severe compression of the fetal heart or great vessels
compression of the normal fetal lung can also rarely cause pulmonary hypoplasia
Differential diagnosis
General imaging differential considerations include:
-
unilocular
does not usually communicate with the bronchial tree, and are therefore typically not air-filled
-
systemic arterial supply
hybrid lesions may present with both CPAM and sequestration features (see above)
-
congenital diaphragmatic herniation
bowel loops within a hemithorax
-
congenital lobar emphysema (congenital lobar overinflation)
hyperlucent and hyperinflated lung segment
no cystic or solid components
localized congenital cystic bronchiectasis
For type I lesions on CT also consider:
cicatrisation collapse with scarring and traction bronchiectasis





 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.