
Pineoblastomas are tumors that are best thought of as small round blue cell tumors located in the pineal region and thus, they closely resemble (both on imaging and on histology) medulloblastomas and retinoblastomas. They are the most aggressive and highest grade tumor among pineal parenchymal tumors and are considered WHO grade 4 tumors 14.
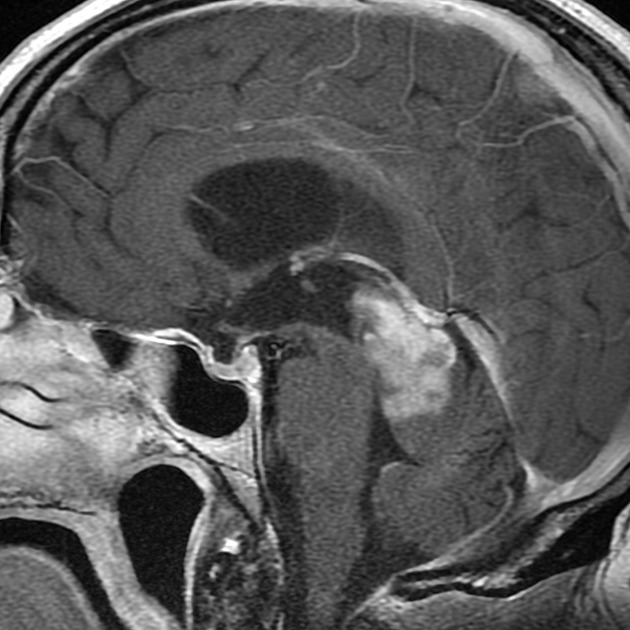
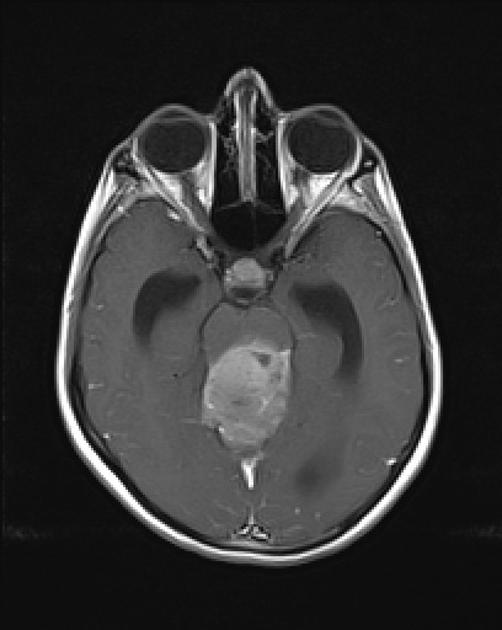
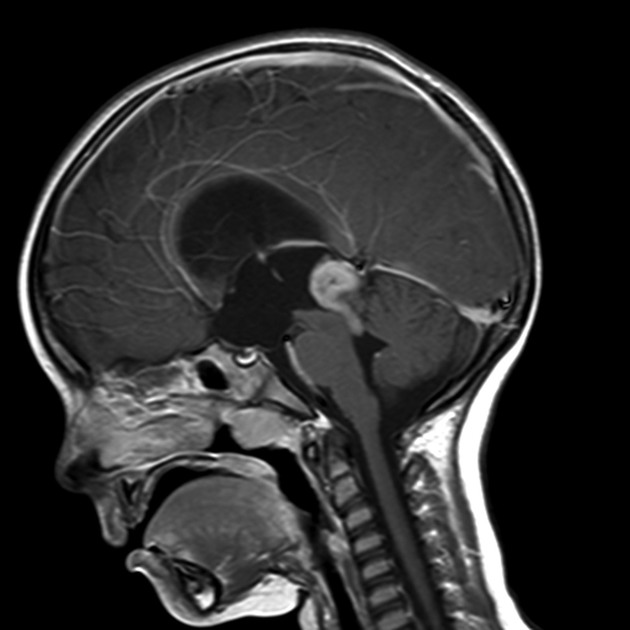
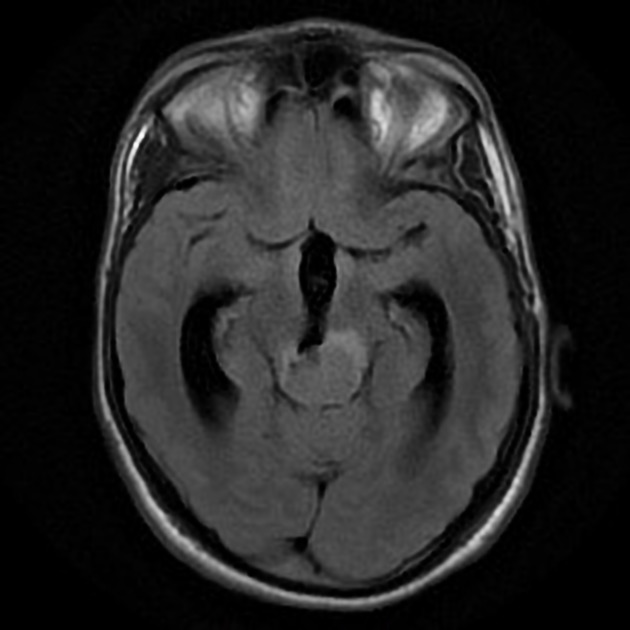
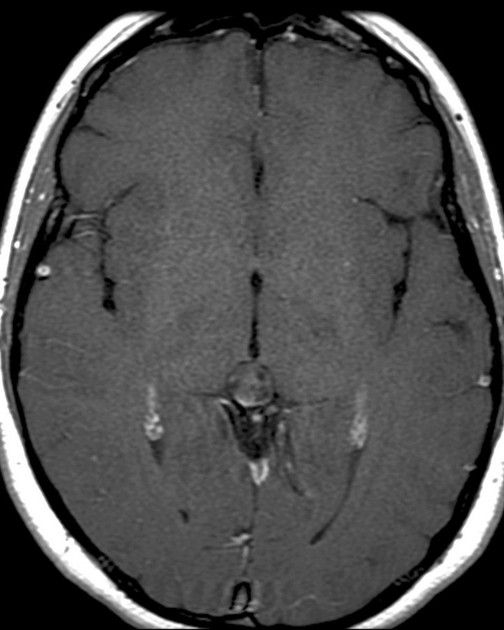
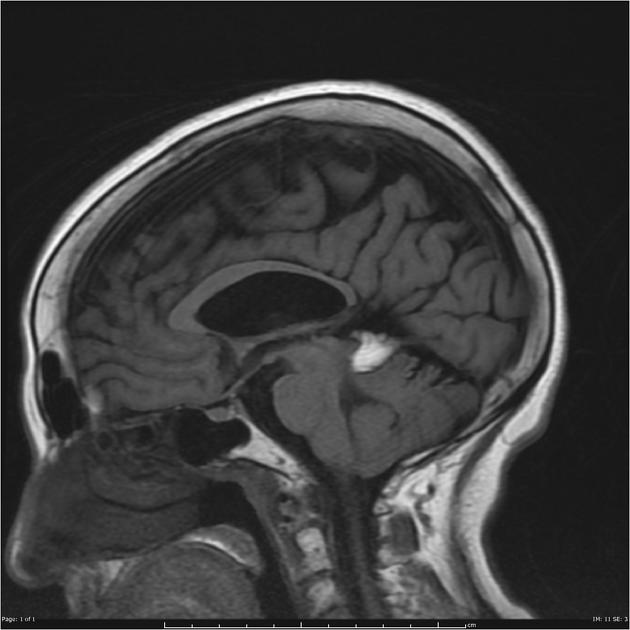
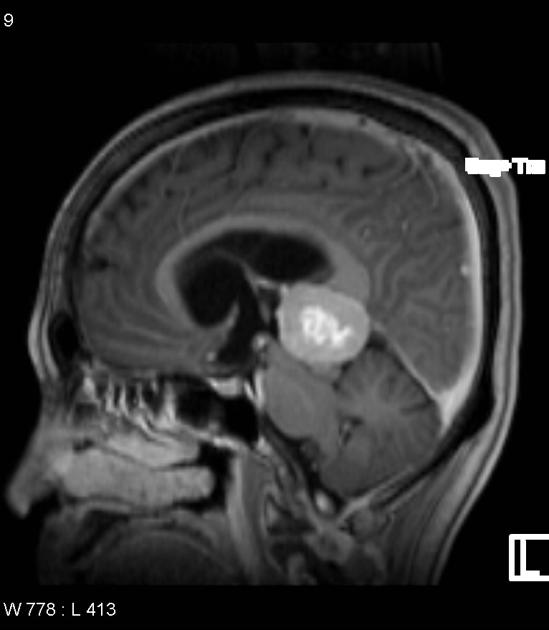
On imaging, they usually present as large lobulated and enhancing tumors (more than 3 cm), hyperattenuating on CT (highly cellular), with heterogeneous signal intensities on MRI, sometimes with evident necrotic and hemorrhage regions. Restricted diffusion is commonly evident and, in almost all cases, obstructive hydrocephalus is observed at the presentation.
On this page:
Epidemiology
Pineoblastomas are the most aggressive pineal parenchymal tumor and account for a substantial proportion of such tumors (24-50%) 7. They are typically found in young children, with only a slight female predilection (M:F 0.7:1; similar to other pineal parenchymal tumors), which is in contrast to the male predominance seen in pineal germinomas) 12.
There is a well-established association with hereditary retinoblastomas. Around 5% of patients with hereditary retinoblastoma (who predominantly have bilateral disease) develop midline (suprasellar or pineal) neuroblastic tumors 6,10. Such cases are sometimes referred to as trilateral retinoblastoma.
Patients with DICER1 syndrome have an increased risk for developing pineoblastomas 13.
Clinical presentation
Pineoblastomas are typically large and almost always associated with obstructive hydrocephalus, due to compression of the cerebral aqueduct. Compression of the tectal plate may also result in the Parinaud syndrome.
They are highly malignant tumors prone to CSF seeding, which is present in 15% of patients at the time of diagnosis.
Pathology
Pineoblastomas originate from pinealocytes and/or their precursors 11. They are the least differentiated pineal cell tumors, with pineocytomas and pineal parenchymal tumor with intermediate differentiation representing better-differentiated tumors along the same spectrum. Pineoblastomas are considered WHO grade 4 tumors 12.
Macroscopic appearance
These tumors are poorly defined, often invading adjacent brain parenchyma. Their cut surface is pink and soft, with areas of necrosis and hemorrhage not infrequently seen 12.
Microscopic appearance
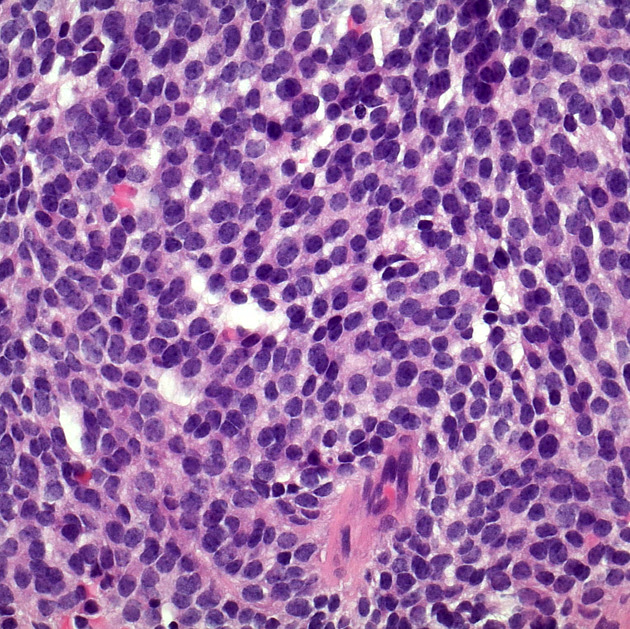
Pineoblastomas are composed of tightly packed small round blue cells (high nuclear to cytoplasmic ratio) which in turn determines their imaging appearances (see below) 7. The growth pattern is largely featureless with the pineocytomatous rosettes seen in pineocytomas not present 12. In contrast, Homer Wright rosettes and Flexner-Wintersteiner rosettes are occasionally seen 12.
Areas of necrosis are frequently encountered 12. Mitotic rate is usually high.
Immunophenotype
Immunophenotype is similar to other pineal parenchymal tumors 12.
synaptophysin: positive
neuron-specific enolase: positive
SMARCB1: positive
other neuronal markers (e.g. neurofilament protein and chromogranin-A): variable
Radiographic features
Pineoblastomas tend to be large poorly defined masses, with frequent CSF seeding at presentation. They have a tendency to involve directly adjacent brain structures, which helps distinguish them from other pineal tumors that tend to be better circumscribed.
CT
The solid component tends to be slightly hyperdense compared to the adjacent brain due to high cellularity. This is a characteristic shared by other small round blue cell tumors such as PNET and medulloblastoma.
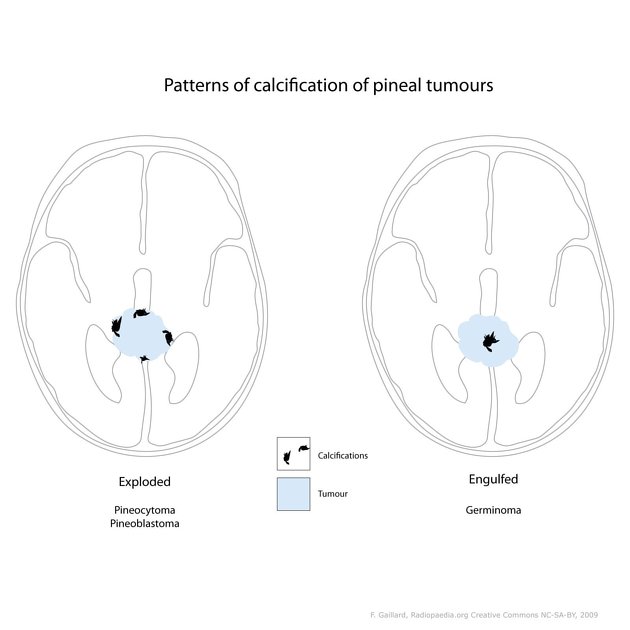
Classically, they are described as having peripherally dispersed or "exploded" calcification (mnemonic: blasted calcification), similar to pineocytomas. In contrast, pineal germinomas tend to engulf pineal calcification.
MRI
Pineoblastomas tend to appear as sizable (>4 cm) irregular masses often with evidence of invasion into the adjacent brain 6,9. Typical signal characteristics include 9:
T1: isointense to hypointense to adjacent brain
-
T2
isointense to adjacent brain
areas of cyst formation or necrosis may be present
T1 C+ (Gd): vivid heterogeneous enhancement
-
DWI/ADC
restricted diffusion due to dense cellular packing
ADC values are typically ~400-800 mm2/s 7
Central necrosis is sometimes present which can make the mass appear centrally cystic and thus can roughly mimic a pineal cyst, although the latter should have a smooth, thin wall 6.
Screening of the whole neural axis is necessary as CSF seeding is seen in 45% of cases 7.
Treatment and prognosis
Treatment is usually a combination of surgery, chemotherapy and radiation 7. Despite treatment, the prognosis has historically been poor, with a 5-year survival as low as 10%. In contrast, in 2021 a 5-year survival of 58-81% has been reported 8,12 with median overall survival times of 4-8 years 12.
The most important factors predicting a favorable outcome are early detection and treatment with at least chemotherapy, preferably a high dose regimen with stem cell rescue 11.
Differential diagnosis
General imaging differential considerations include:
-
other pineal parenchymal tumors
pineocytoma: mature well-differentiated tumor: smaller and better circumscribed
-
thin (<2 mm) wall
astrocytoma of the pineal gland
-
imaging is very similar
located in the vermis rather than pineal region but can be difficult to distinguish if very high in the vermis and very large



 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.