
Posterior cortical atrophy, also known as Benson syndrome, is an uncommon neurodegenerative disease typically affecting individuals in the sixth and seventh decades of life. It is characterized by dysfunction of the parietal, posterior temporal, and occipital lobes which results in progressive apraxias, problems with visuospatial and visuoperceptual perception as well as literacy deficiencies 4.
On this page:
Terminology
As is the case with many neurological diseases, the literature is replete with variable terminology. Although generally thought of as a variant of Alzheimer disease (and thus sometimes referred to as a visual variant of Alzheimer disease) it is now recognized as sometimes being caused by other pathologies (see below) 4. As such posterior cortical atrophy should be considered a clinical syndrome with variable etiology.
Epidemiology
Posterior cortical atrophy is generally diagnosed in the sixth and seventh decades (50-65 years of age) with no sex predilection 4.
Incidence is unknown largely in part to the lack of general awareness of the condition and lack of consistent diagnostic criteria. As such the entity is likely underrecognized 4.
Clinical presentation
Posterior cortical atrophy is clinically dominated by disruption of normal higher-order visual processes, and as such patients eventually behave like individuals who are blind. Patients typically present with 4:
visual agnosia: early and pronounced feature
apraxia: early and pronounced feature
alexia (difficulty reading)
environmental disorientation
-
left/right disorientation
various positive perceptual phenomena e.g. reverse size phenomena, upside-down phenomena etc.
Approximately 25% of patients with posterior cortical atrophy will also develop visual hallucinations 1. These patients may represent a distinct subgroup, with hallucinations believed to be due to the complex interplay between the midbrain, thalamus and primary visual cortex, rather than the visual association areas 1.
Although mild memory impairment is often present early in the disease, it is clinically different from that seen in Alzheimer disease.
Pathology
In most individuals with posterior cortical atrophy, histology demonstrates focal degeneration with the presence of neuritic plaques and neurofibrillary tangles, especially in parieto-occipital and temporo-occipital areas, and less commonly in the primary visual cortex 4. As such, posterior cortical atrophy shares features with Alzheimer disease, so much so that some authors believe it to be a variant of the latter, and refer to it as a visual variant of Alzheimer disease. Indeed pathologically the conditions are essentially indistinguishable, other than the distribution of pathological changes and age of onset.
It should be noted, that in some individuals who present with posterior cortical atrophy, other underlying etiologies are identified including dementia with Lewy bodies, corticobasal degeneration and even prion disease 4.
Furthermore, a number of gene mutations have been identified in posterior cortical atrophy including presenilin 1 and 2 genes (PSEN1 and PSEN2), prion protein gene (PRNP), progranulin gene (GRN) and microtubule-associated protein tau gene (MAPT) 5,6.
Radiographic features
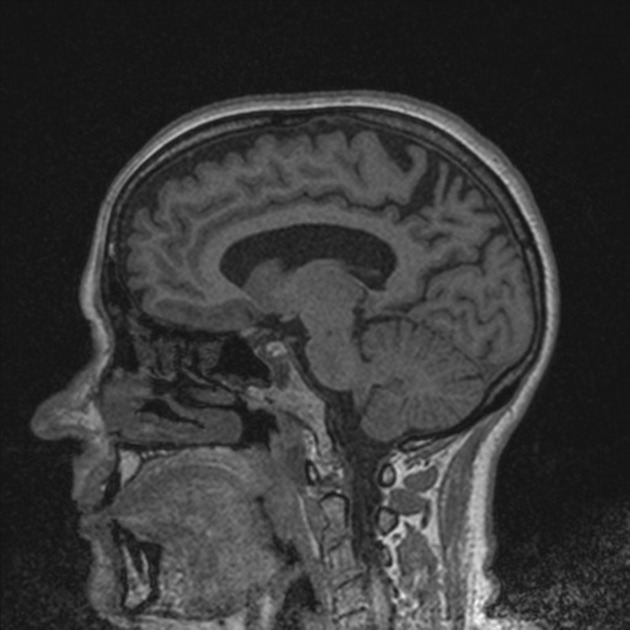
MRI is the modality of choice for assessing patients with neurodegenerative diseases, although CT may allow gross volume changes to be appreciated. Nuclear medicine functional studies are also of benefit 4.
MRI
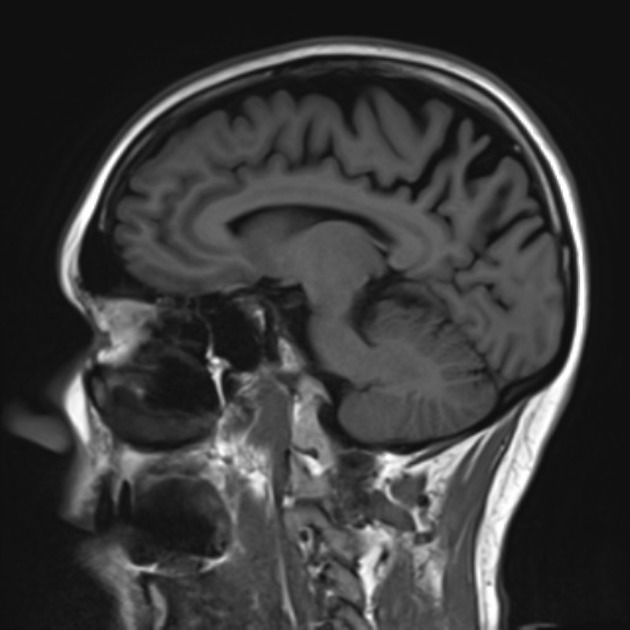
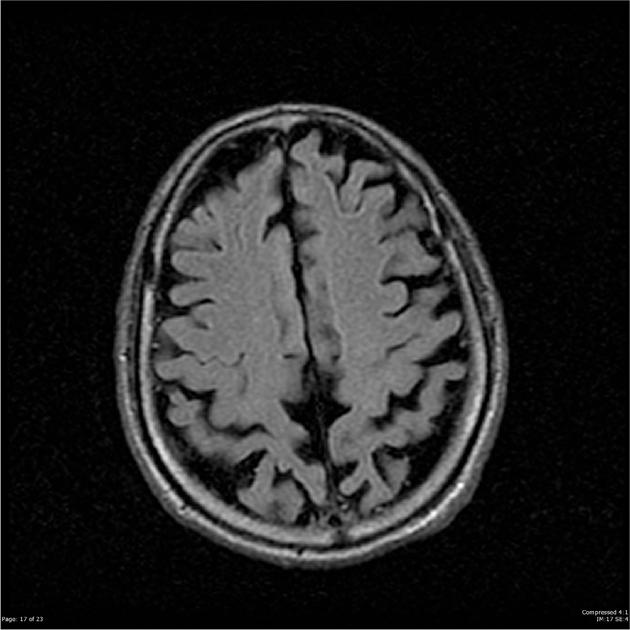
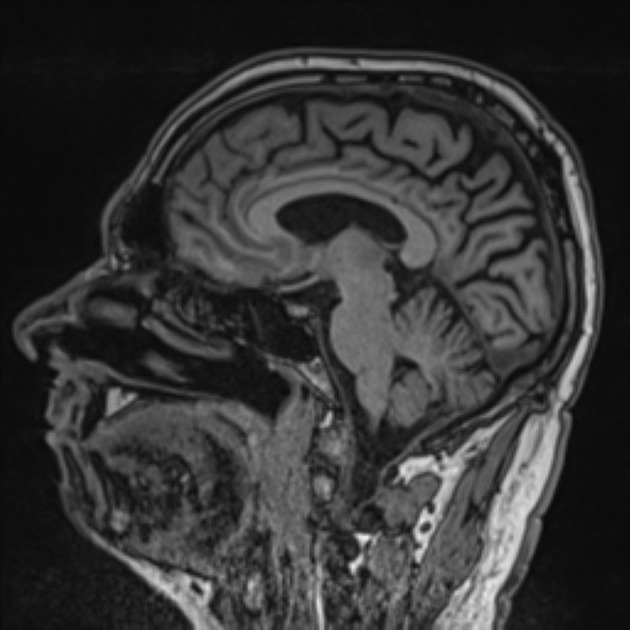
The main findings are bilateral, but often more pronounced right-sided, parietal and parieto-occipital and temporo-occipital atrophy. Hippocampi are relatively normal.
Parieto-occipital sulcus is reported to be wider in PCA compared to AD7.
Nuclear medicine
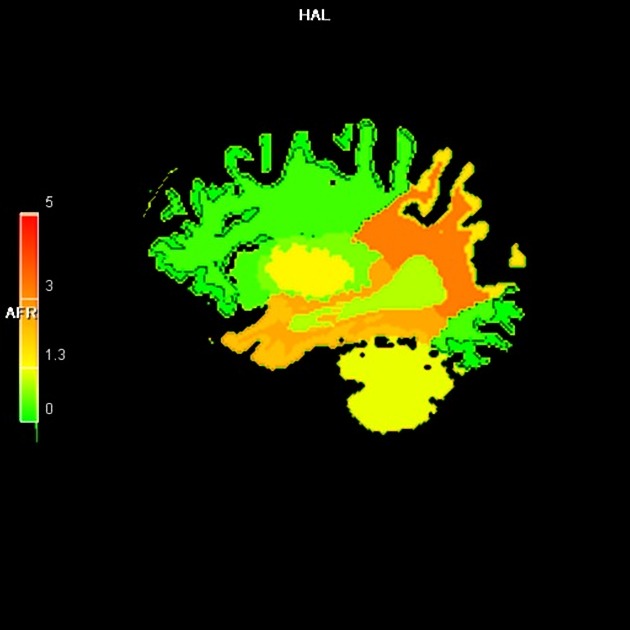
SPECT and PET demonstrate hypoperfusion and hypometabolism in the same areas as affected by atrophy and these changes may precede morphological change 4.
Treatment and prognosis
As is the case with most neurodegenerative diseases, no cure is available. Management is medical and centers on behavioral techniques targeted at overcoming visual disabilities as well as the use of antidepressants.
The disease is gradually progressive with patients usually succumbing within 8-12 years from the time of symptom onset.
History and etymology
Posterior cortical atrophy was first described by Franck D Benson in 1988 and thus is also known as Benson syndrome 2.
Differential diagnosis
The primary differential diagnosis includes:
-
older age at onset
more pronounced memory loss as an early feature
visuospatial features not prominent
-
frontal lobes and basal ganglia/thalami/midbrain atrophy
Parkinsonian symptoms (later in the disease)
-
Creutzfeldt-Jakob disease (CJD)
can have a very similar presentation
clinically CJD has a more rapid time course


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.