
Pilocytic astrocytomas, also known as juvenile pilocytic astrocytomas, are circumscribed astrocytic gliomas that typically occur in young patients. The majority of sporadic pilocytic astrocytomas arise from the cerebellum, whereas in the setting of neurofibromatosis type 1 (NF1), they often involve the optic chiasm and pathway. In adults, they are typically supratentorial 11.
Pilocytic astrocytomas are considered WHO grade 1 tumors in the WHO classification of CNS tumors and correspondingly have a relatively good prognosis 6.
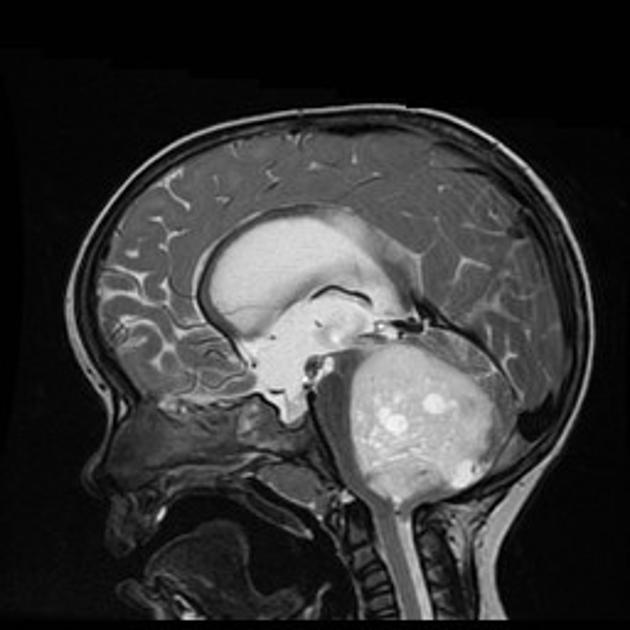
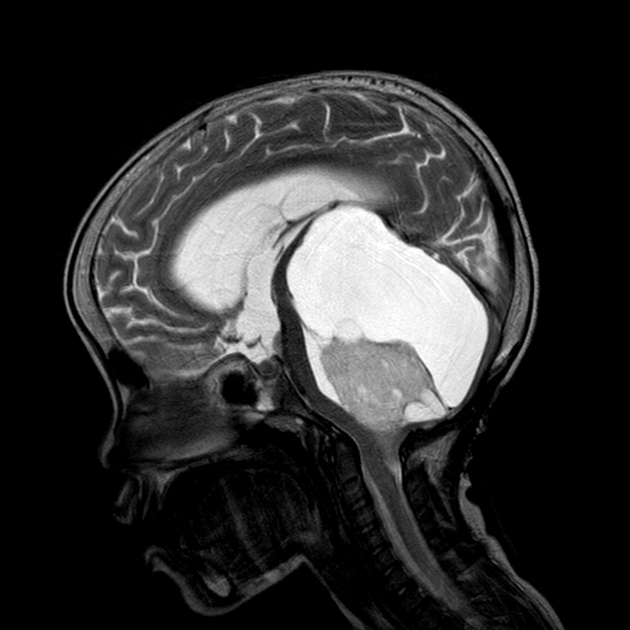
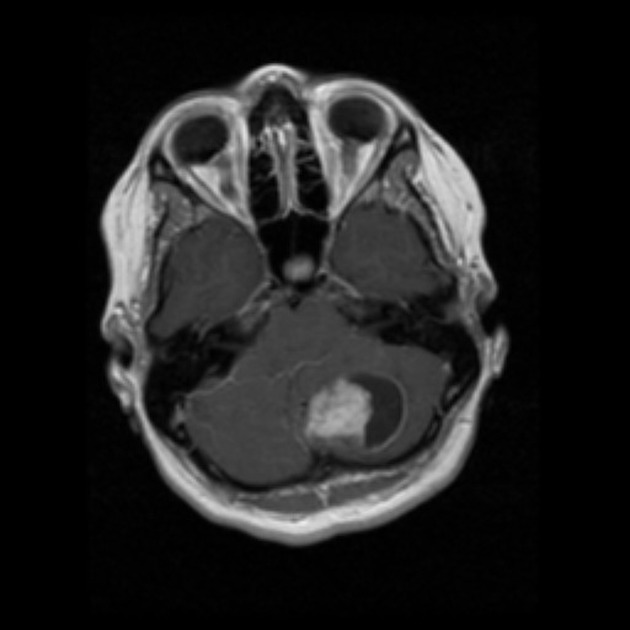
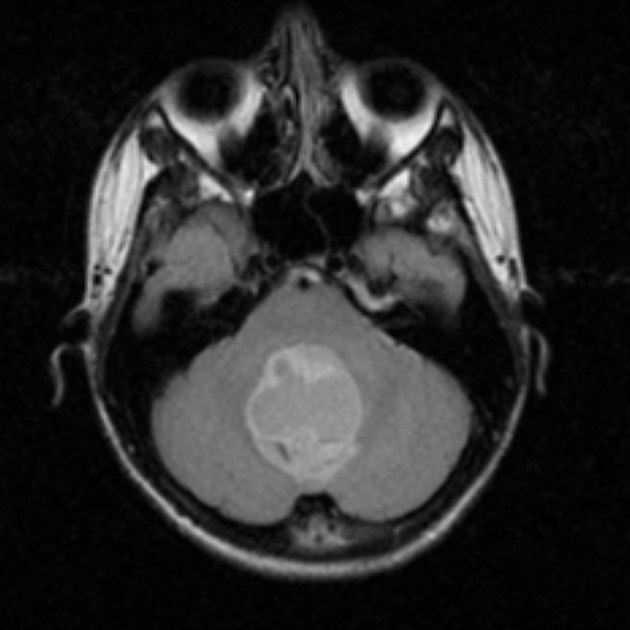
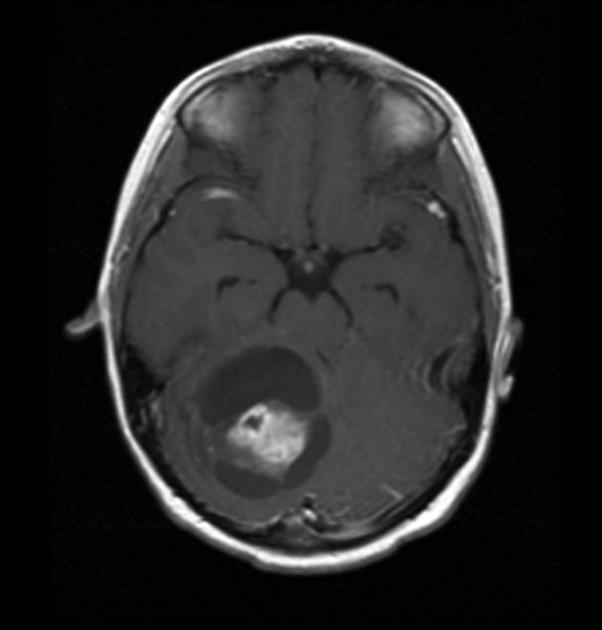
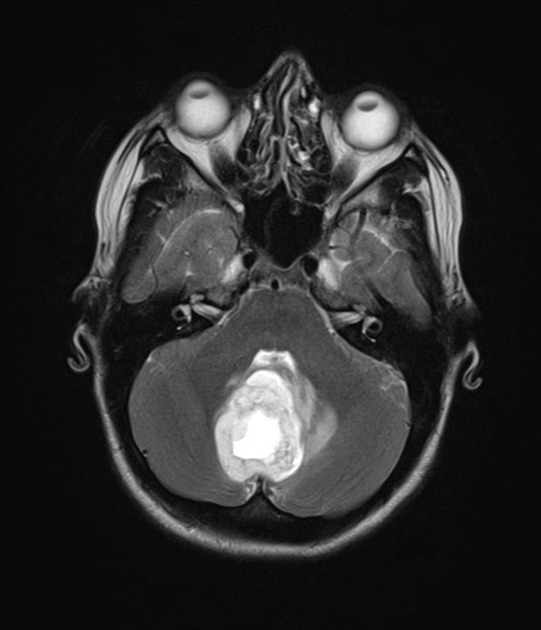
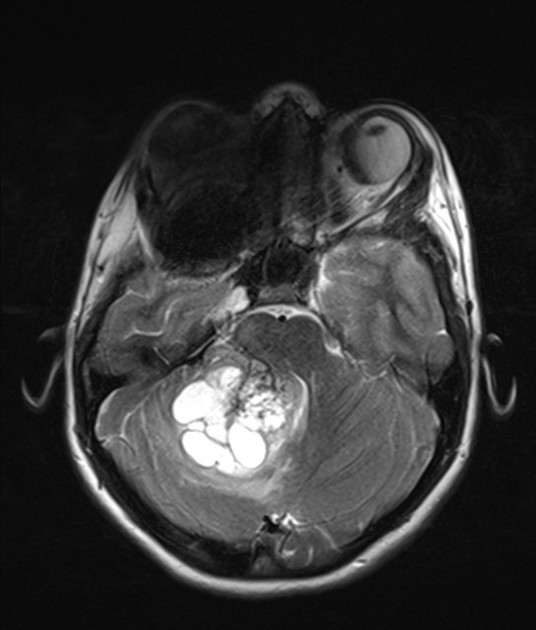
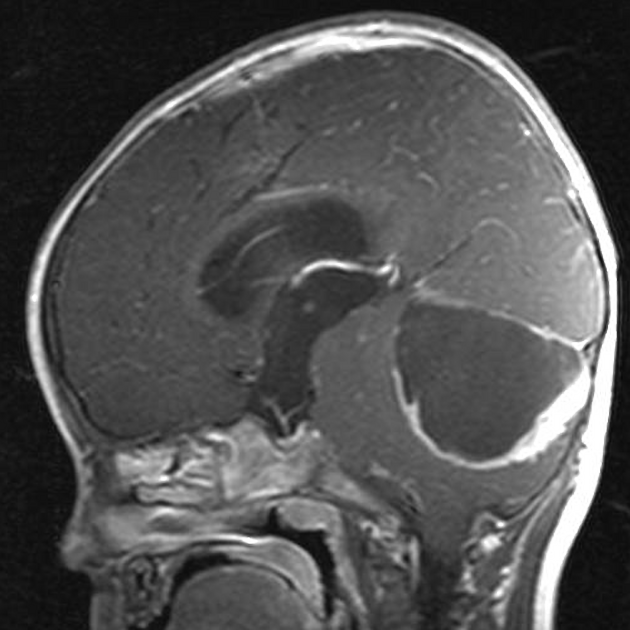
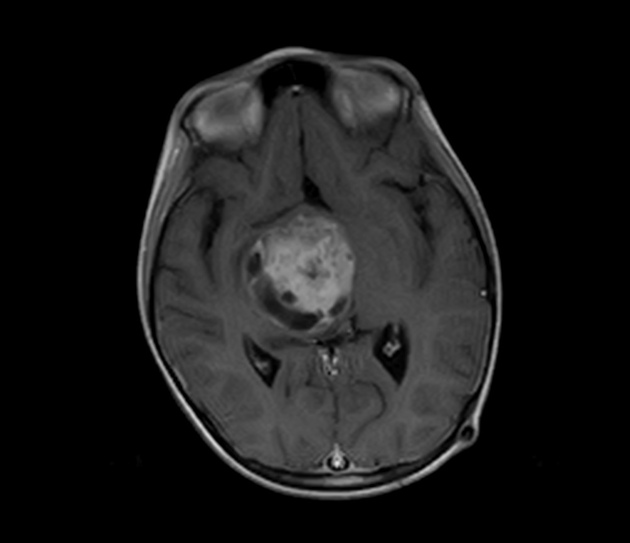
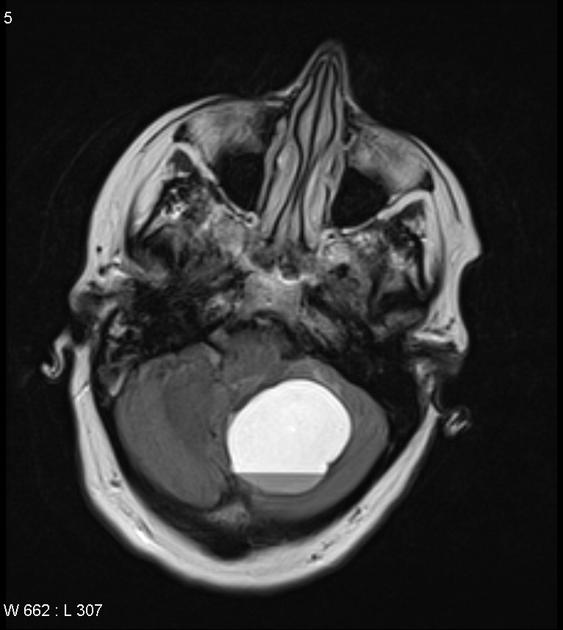
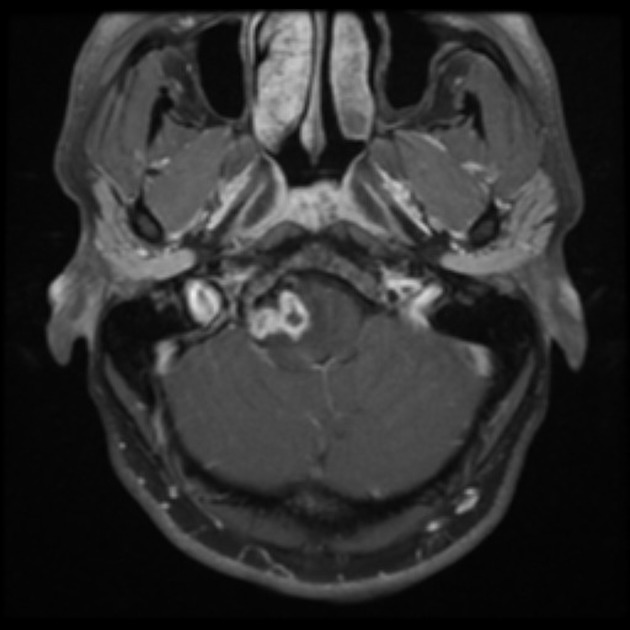
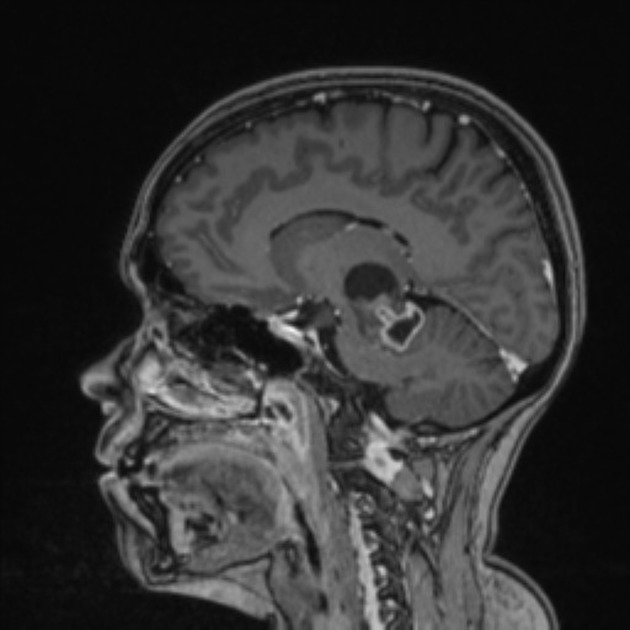
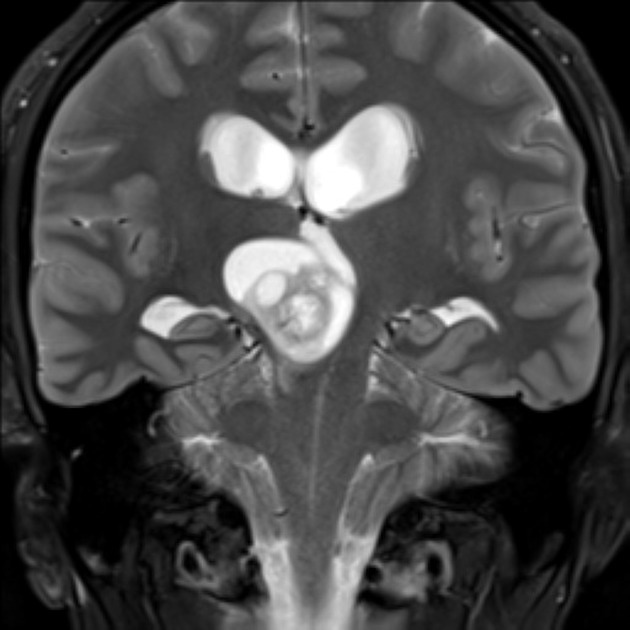
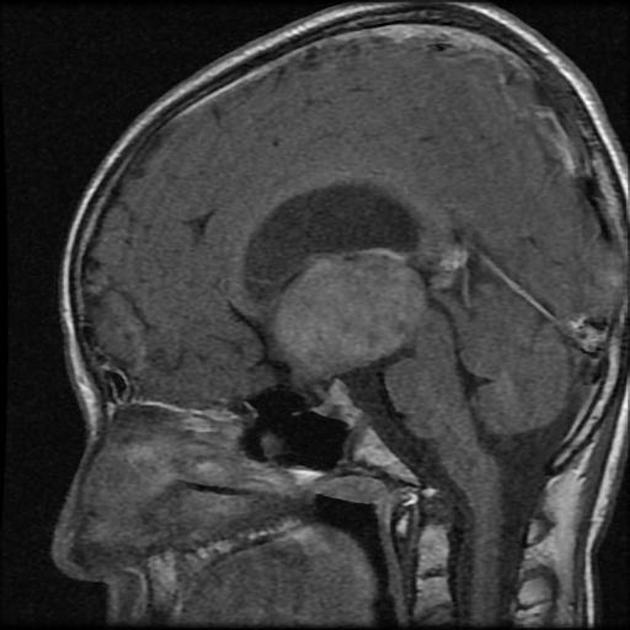
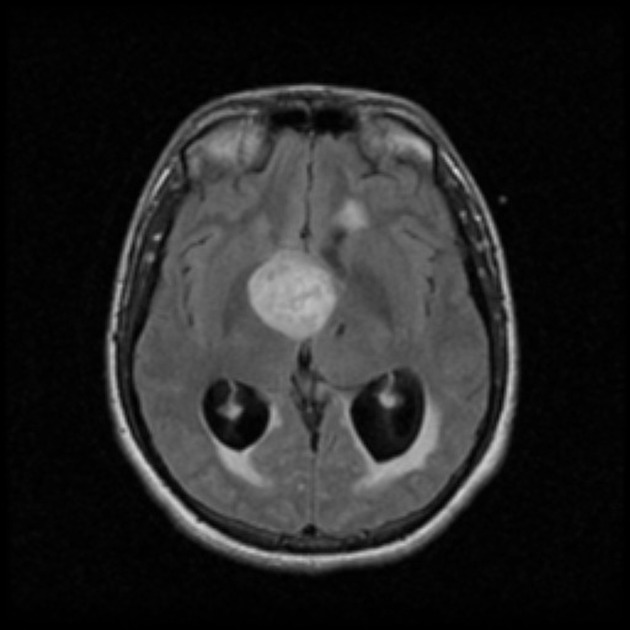
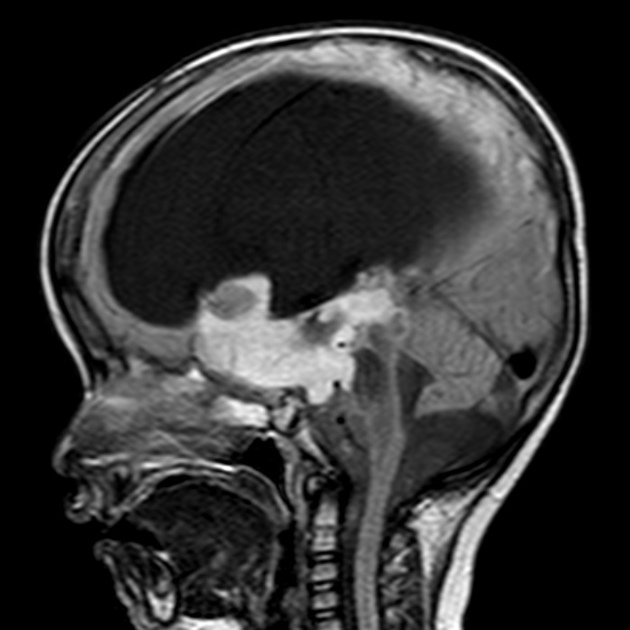
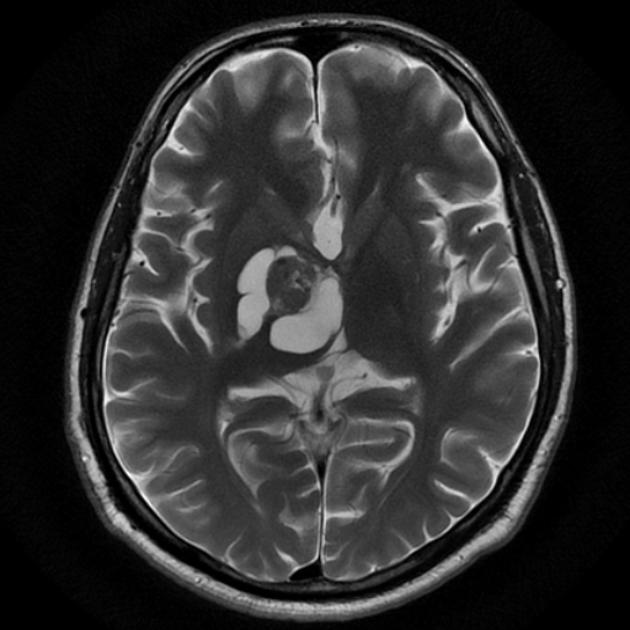
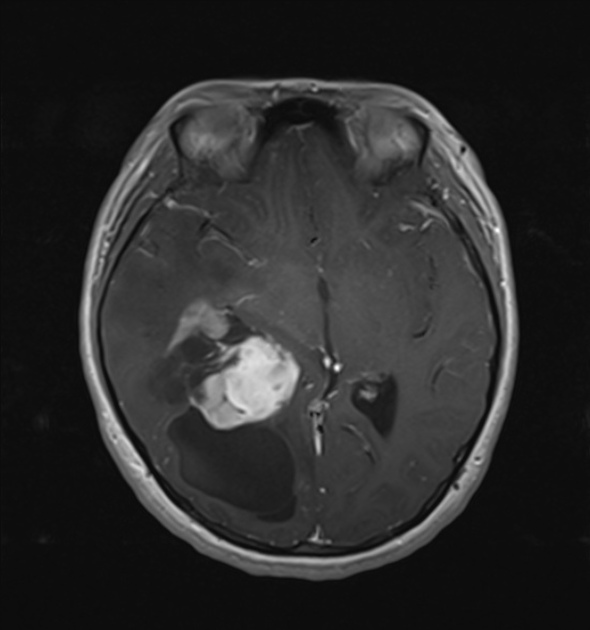
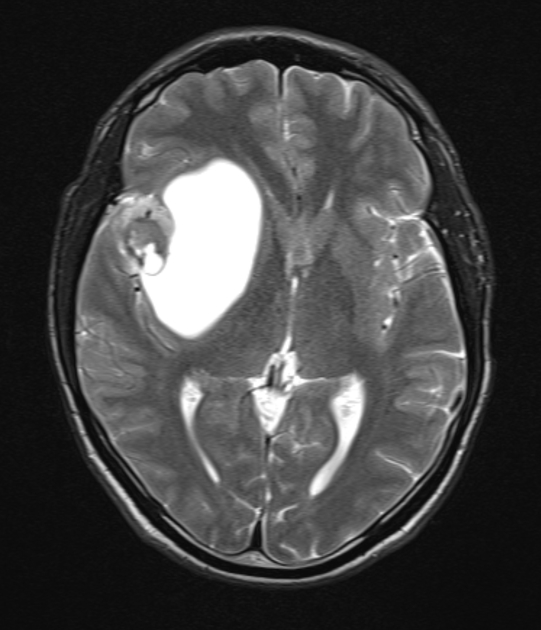
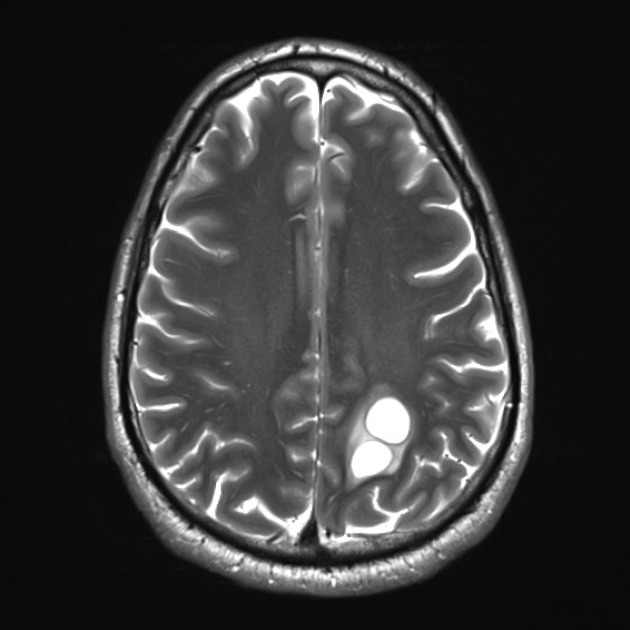
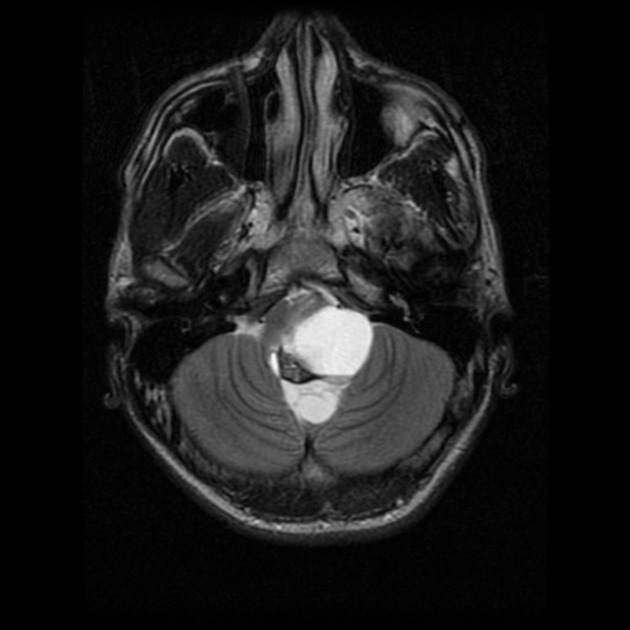
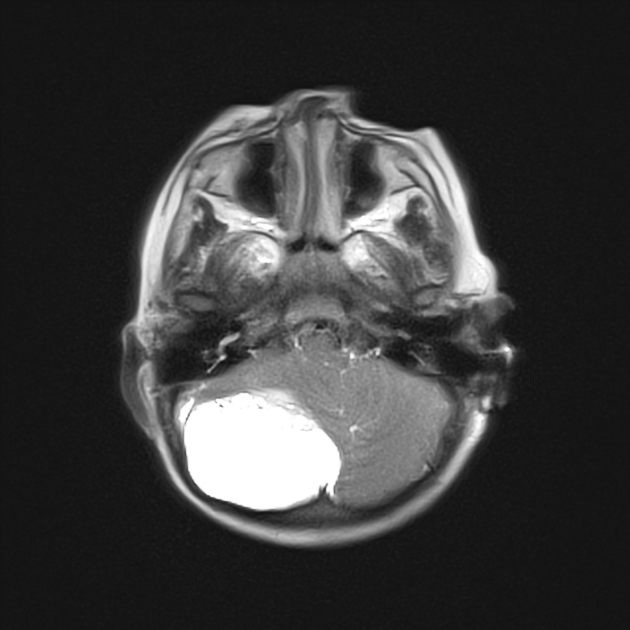
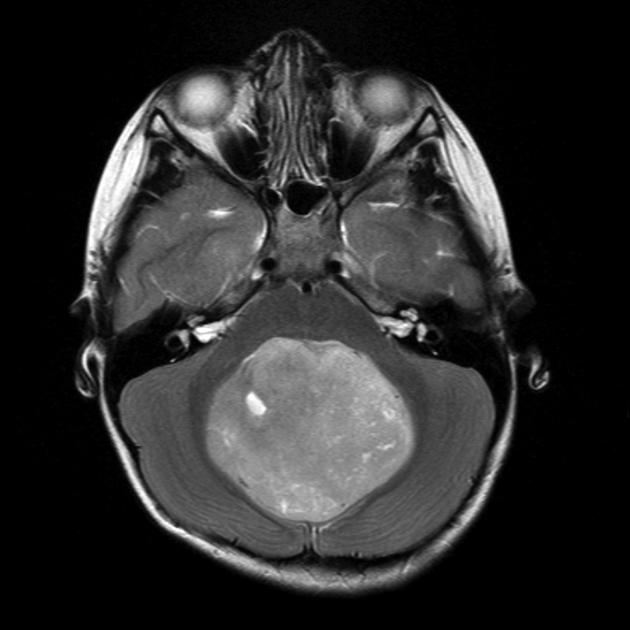
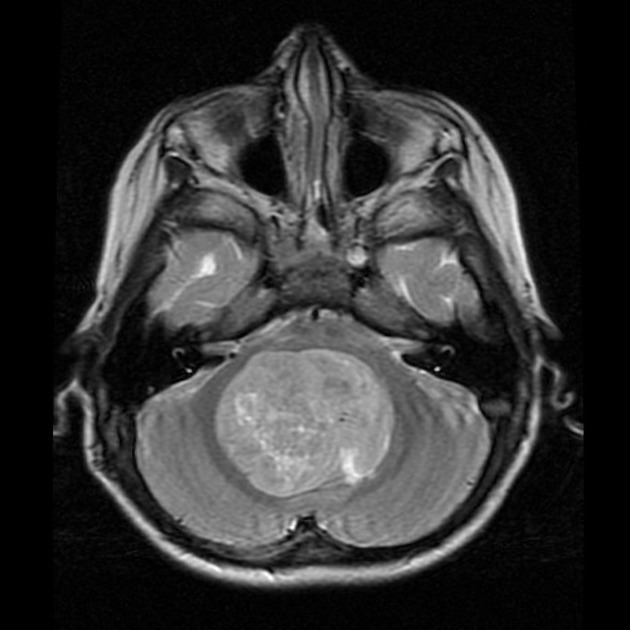
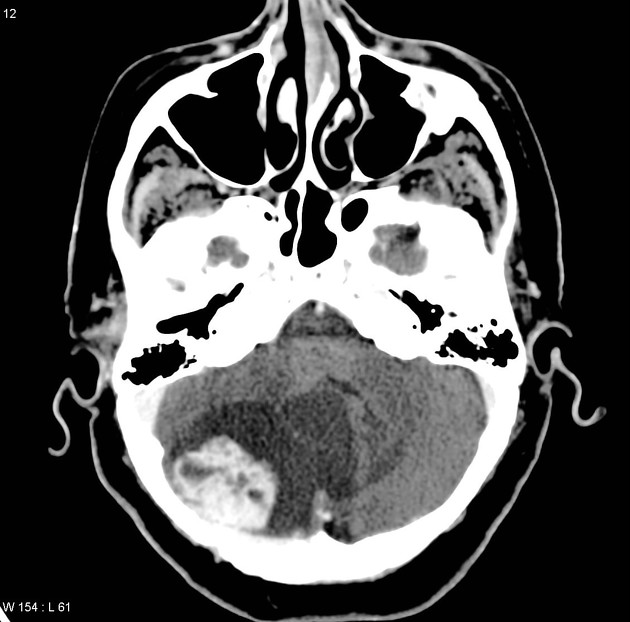
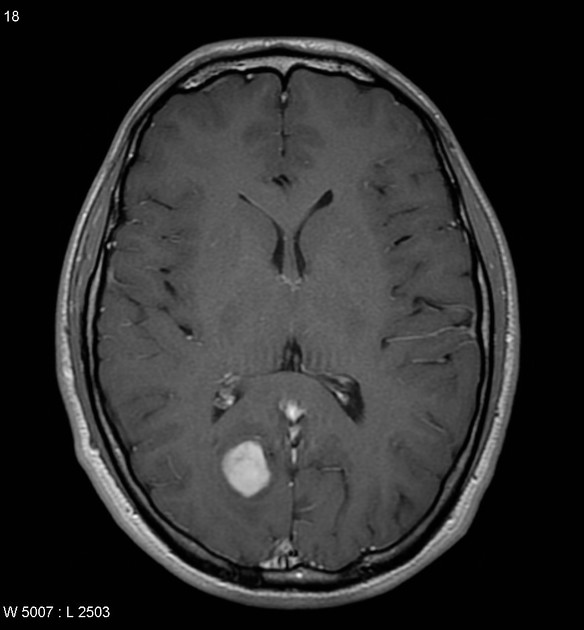
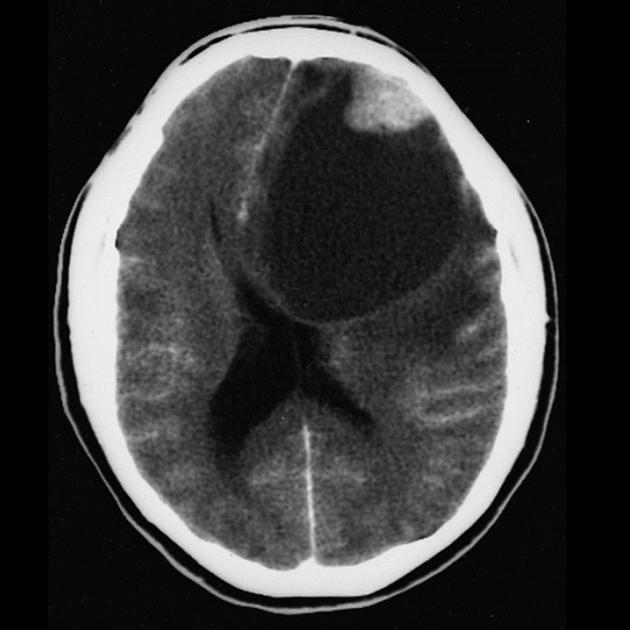
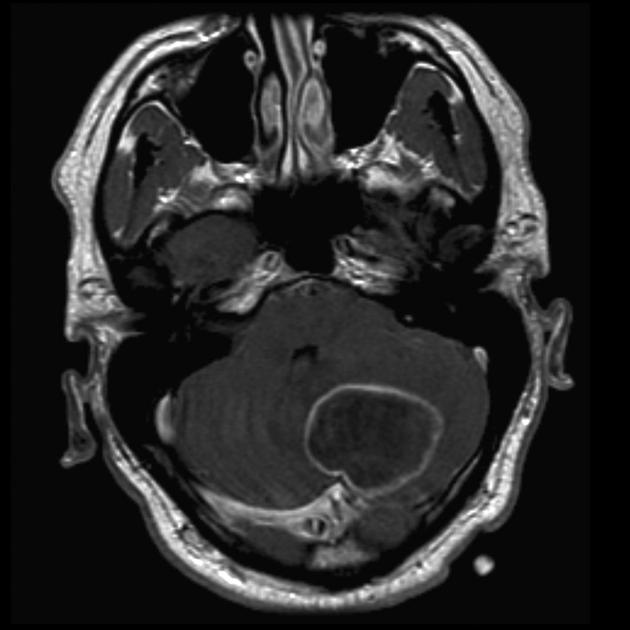
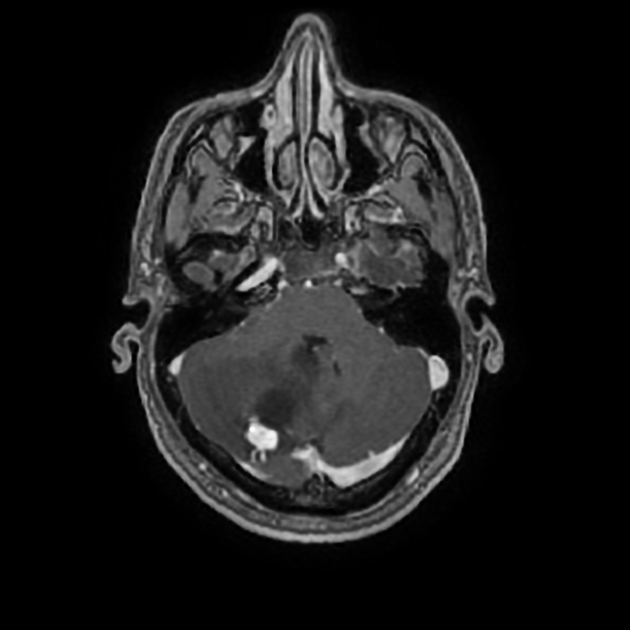
These tumors have a range of imaging appearances, with the majority presenting as a large cystic lesion with a brightly enhancing mural nodule. Calcification can be present in around one-fifth of cases.
Optic pathway gliomas and spinal cord pilocytic astrocytomas are discussed separately. The remainder of this article focuses on a general discussion of pilocytic astrocytomas, particularly those in the cerebellum.
On this page:
Epidemiology
Pilocytic astrocytomas are tumors of young people, with ~70% occurring in the first two decades of life, typically late in the first decade (9-10 years) 11. They do, however, also occasionally arise in adults 11.
Although they account for ~2.5% (range 0.6-5.1%) of all intracranial neoplasms (~4% of all glial tumors), they are the most common primary brain tumor of childhood (~15%) 7 and the second most common pediatric posterior fossa tumor (~30%) after medulloblastomas 10.
Associations
There is a strong association with NF1. NF1-associated tumors have a tendency to affect the optic nerves and chiasm (see: optic pathway glioma). The association between NF1 and pilocytic astrocytomas is so strong that up to 20% of all patients with NF1 will develop these tumors, typically in early childhood. Conversely, approximately one-third of pilocytic astrocytomas involving the optic nerves have associated NF1.
Clinical presentation
Presentation depends on location. In posterior fossa tumors, there is predominantly a mass effect with signs of raised intracranial pressure, especially when hydrocephalus is present. Bulbar symptoms or cerebellar symptoms may also be present.
Pathology
The cell of origin of pilocytic astrocytomas has not been established and may be different depending on location. This is supported by the DNA methylation profile of infratentorial and supratentorial pilocytic astrocytomas being distinct but shared with region-specifc astrocytes and neuronal stem cells 15,16.
Grading
All pilocytic astrocytomas are considered WHO CNS grade 1 tumors, eventhough some have higher grade features including elevated mitotic activity and/or necrosis. These can be referred to as pilocytic astrocytoma with histological features of anaplasia, and may have a worse prognosis although this has not yet been fully determined 6.
Pilomyxoid astrocytoma is considered a subtype and is more aggressive. High-grade astrocytoma with piloid features is now considered a separate entity 6.
Location
By far the most common location is the cerebellum, with the optic pathway being the next most common, particularly in patients with NF1. In adults, most tumors are supratentorial 11.
The distribution within the cerebellum varies with many tumors involving both the vermis and the cerebellar hemisphere.
In general, pilocytic astrocytomas typically arise from midline structures.
-
cerebellum
60%
-
optic pathway (optic nerve, optic chiasm, hypothalamus, optic radiation)
25-30%, particularly common in NF1
-
other less common locations
brainstem
cerebral hemispheres: more frequent in adults 11
cerebral ventricles
velum interpositum
spinal cord: see spinal pilocytic astrocytoma
Macroscopic appearance
Pilocytic astrocytomas vary significantly in morphology ranging from tumors dominated by a large cyst with a smaller mural nodule, to mostly solid timours, and particularly in adults, heterogenous masses with hemorrhage, necrosis and calcification.
The classic appearing is, however, of a cyst with a mural nodule, in many ways similar to hemangioblastomas. The cyst is believed to the result of increased expression of vascular endotherlial growth factor (VEGF) which not only promotes angioneogenesis but also increases vascular permeability 14.
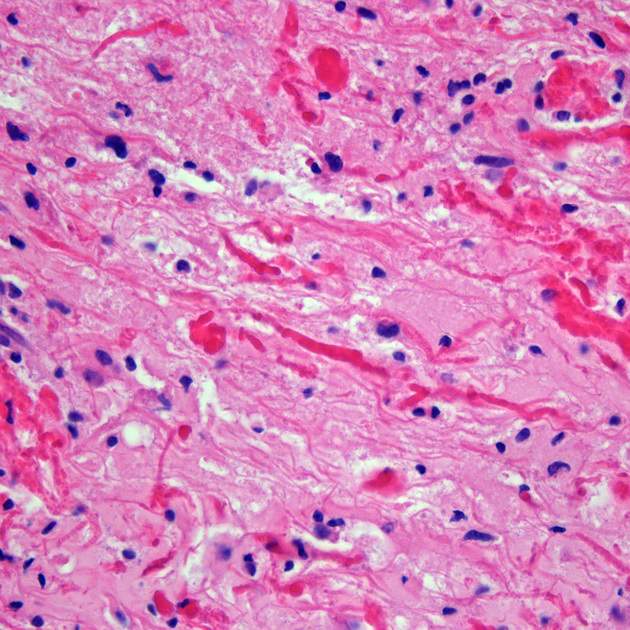
Microscopic appearance
The term pilocytic refers to the elongated hair-like projections from the neoplastic cells 4. The presence of eosinophilic Rosenthal fibers is a characteristic feature and hyalinization of blood vessels is also common. The histological features are, however, fairly heterogeneous even within the one tumor, with some areas mimicking diffuse astrocytomas and even oligodendrogliomas 6.
Immunophenotype
Immunohistochemistry reflects astrocytic differentiation 6:
GFAP: positive
S100: positive
OLIG2: positive
IDH R132H mutation: negative
p53 protein: negative or weak
Genetics
Pilocytic astrocytoma almost invariably have alterations in the MAPK pathway. In patients with neurofibromatosis type 1, the mutations are in the NF1 gene. Otherwise, BRAF alterations are most common, (present in ~70% of cases). Other relatively common mutations include FGFR1 mutations or fusion and KRAS mutations 6. Importantly they, along with other pediatric low-grade gliomas, lack IDH mutations and TP53 mutations 6,7.
Radiographic features
Pilocytic astrocytomas range in appearance:
-
large cystic component with a brightly enhancing mural nodule: 67%
non-enhancing cyst wall: 21%
enhancing cyst wall: 46%
heterogeneous, mixed solid and multiple cysts and central necrosis: 16%
completely solid: 17%
Enhancement is almost invariably present (~95%). Up to 20% may demonstrate some calcification. Hemorrhage is an uncommon complication.
MRI
-
T1
solid component: iso- to hypointense compared to adjacent brain
cystic component: fluid signal unless hemorrhagic
-
T1 C+ (Gd)
vivid contrast enhancement
the cyst wall enhances in ~50% of cases
enhancement does not necessarily imply presence of tumor cells, particularly if very thin 2,6,9
-
T2
solid component: hyperintense compared to adjacent brain
cystic component: high signal
T2*/GRE/SWI: signal loss if calcification or hemorrhage present
DWI/ADC: usually facilitated diffusion (DWI restriction with low ADC has been reported in BRAF V600E tumors13)
MRS: high choline and lactate peak13
Treatment and prognosis
They are slow-growing well-circumscribed tumors with an overall good prognosis in children following treatment (5-year and 10-year survival >95%) 6. Cystic tumors have an even better prognosis while fibrillary variants tend to do worse.
Surgical resection, if complete, is usually curative. Some surgeons advocate that only the nodule needs to be resected to achieve a cure, as the cyst walls are non-neoplastic, even if enhancing 2,6. Other studies suggest that the likelihood of tumor being present in the cyst wall is much higher if enhancement is present and if often obvious intra-operatively 9.
Adults have a poorer prognosis, with a ~50% 5-year survival 11,12.
It is also worth noting that historically some patients with neurofibromatosis type 1 and high-grade cerebellar tumors diagnosed as pilocytic astrocytomas, actually represented high-grade astrocytoma with piloid features.
History and etymology
In 1931, Harvey Cushing was the first who described this tumor based on his studies of 76 cases of cerebellar astrocytomas 5. Pilocytic means "hair-like" and is derived from the Latin word pilus for hair.
Differential diagnosis
General imaging differential considerations include:
-
high-grade astrocytoma with piloid features
particularly in adults and in patients with neurofibromatosis type 1
terrible prognosis
-
usually seen in adults. In children, usually associated with von Hippel-Lindau disease
cyst wall usually does not enhance
doesn't show calcifications
smaller mural nodule with angiographic contrast blush
-
typically arise from the midline (especially the vermis and roof of the fourth ventricle) rather than the cerebellar hemisphere
usually seen in younger patients (2-6 years of age)
-
atypical teratoid/rhabdoid tumor (AT/RT)
larger heterogeneously enhancing mass
-
tends to fill the fourth ventricle and protrude out of the foramina of Luschka and foramen of Magendie
large cystic component less common




 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.