
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig disease and Charcot disease, is the most common form of motor neuron disease 1,4 resulting in progressive weakness and eventual death due to respiratory insufficiency. There is both upper motor neuron and lower motor neuron damage. 'Lateral sclerosis' indicates degeneration of the pyramidal tracts.
On this page:
Epidemiology
Amyotrophic lateral sclerosis typically is diagnosed in middle age. There is a recognized male predilection 1.
Clinical presentation
Both upper and lower motor neurons are affected, with decreased motor strength and wasting of the limb muscles, bulbar muscles, and diaphragm. There is a progressive loss of motor strength, with preservation of intellectual and sensory function. In the hands, the split hand sign or split hand plus sign may be characteristically seen.
El Escorial criteria for the diagnosis of amyotrophic lateral sclerosis 7:
-
it requires the presence of
signs of lower motor neuron (LMN) degeneration by clinical, electrophysiological or neuropathologic examination
signs of upper motor neuron (UMN) degeneration by clinical examination
progressive spread of signs within a region or to other regions
-
together with the absence of
electrophysiological evidence of other disease processes that might explain the signs of LMN and/or UMN degenerations
neuroimaging evidence of other disease processes that might explain the observed clinical and electrophysiological signs
Variants
There are many clinical variants of amyotrophic lateral sclerosis, the most common being:
Rarer variants include:
flail arm syndrome (brachial amyotrophic diplegia or Vulpian-Bernhardt syndrome)
flail leg syndrome
hemiplegic amyotrophic lateral sclerosis (Mills syndrome)
O’Sullivan-McLeod syndrome
pseudopolyneuritic amyotrophic lateral sclerosis (Patrikios disease)
facial onset sensory and motor neuronopathy
finger extensor weakness with downbeat nystagmus-motor neuron disease
Pathology
Amyotrophic lateral sclerosis is a relentlessly progressive neurological disorder characterized by the death of upper motor neurons (Betz cells in the cortex) and anterior horn cells with secondary Wallerian degeneration 2.
Genetics
The majority of cases are sporadic and thus less well understood. In the familial form of amyotrophic lateral sclerosis, and in some sporadic cases, several gene mutations have been identified (e.g. the hexanucleotide repeat expansion in C9orf72 (most common familial cause), SOD1, TDP-43, FUS) 5.
Radiographic features
MRI
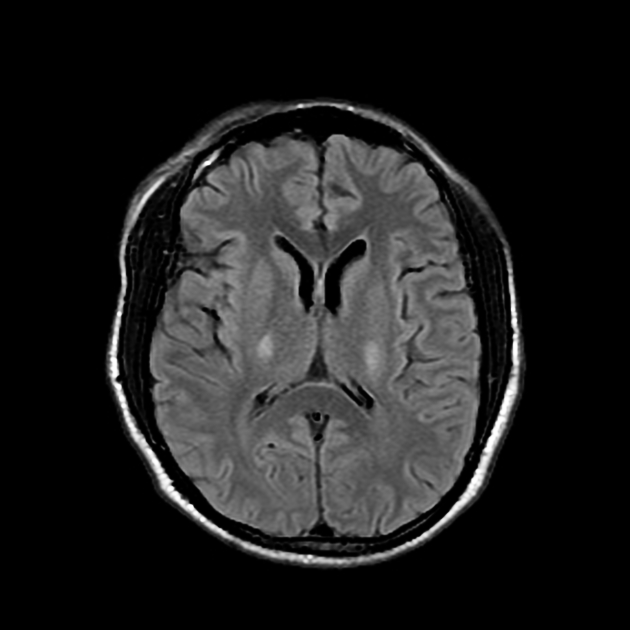
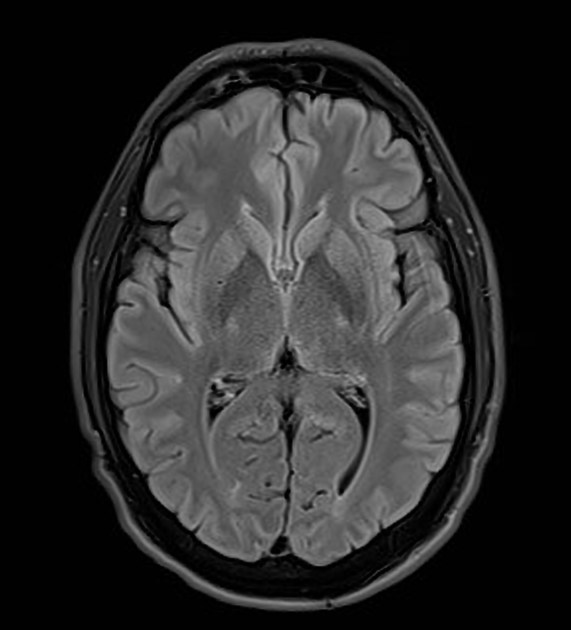
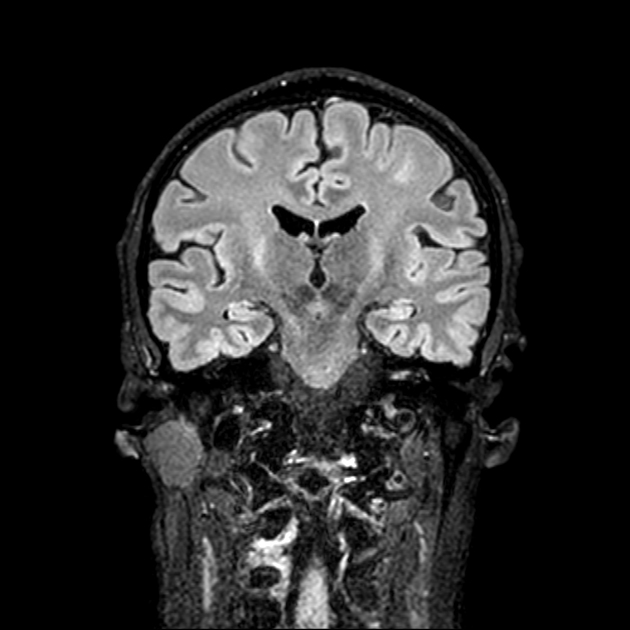
MRI of the neuraxis in amyotrophic lateral sclerosis may be normal. Potential abnormalities include:
T1: hyperintensity of the tongue may be seen in patients with bulbar involvement, known as the "bright tongue sign" 13
-
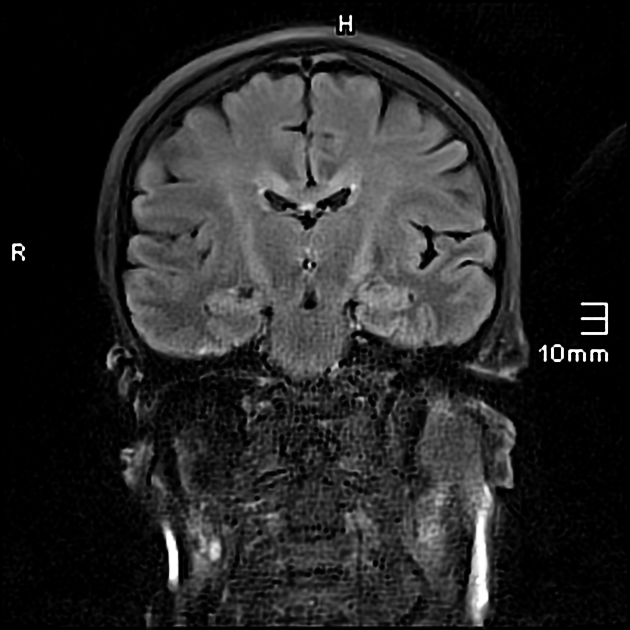
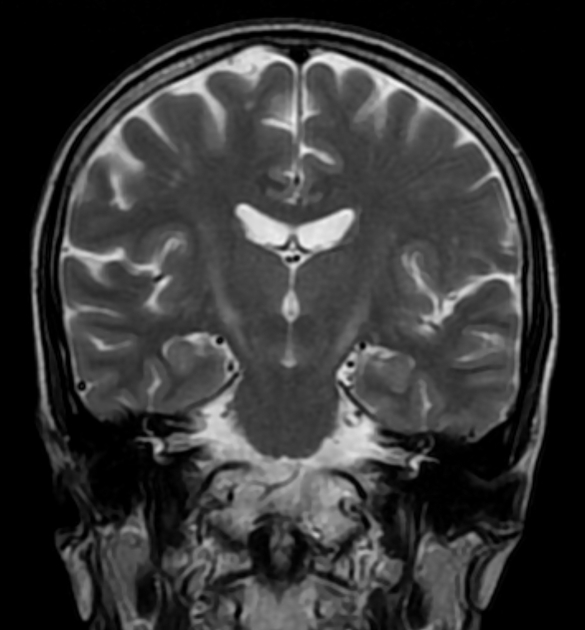
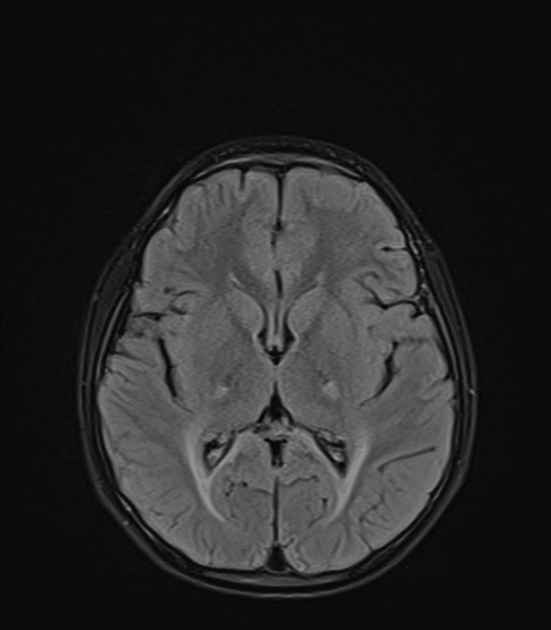
T2: hyperintensity in the corticospinal tracts
seen earliest in the internal capsule, as the fibers are most concentrated here
eventually, the entire tract from motor strip to the spinal cord is affected by increased T2 signal and volume loss 3
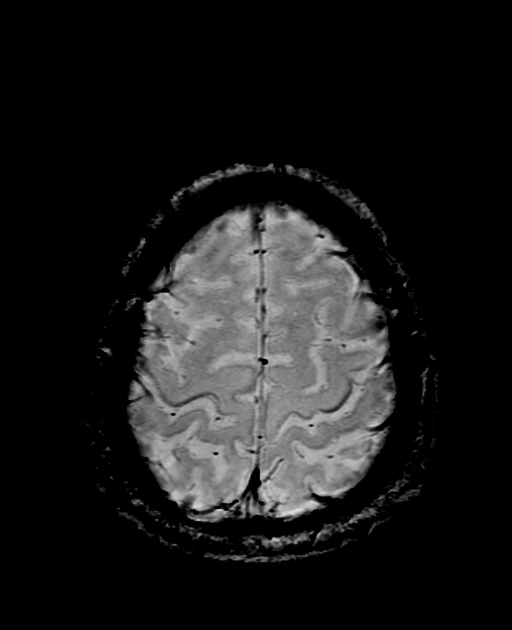
despite this being a well-recognized radiological feature of amyotrophic lateral sclerosis, corticospinal tract T2 hyperintensity is only seen in 30% of cases 14
the sensitivity and specificity are rather low: specificity <70% and sensitivity <40% 6
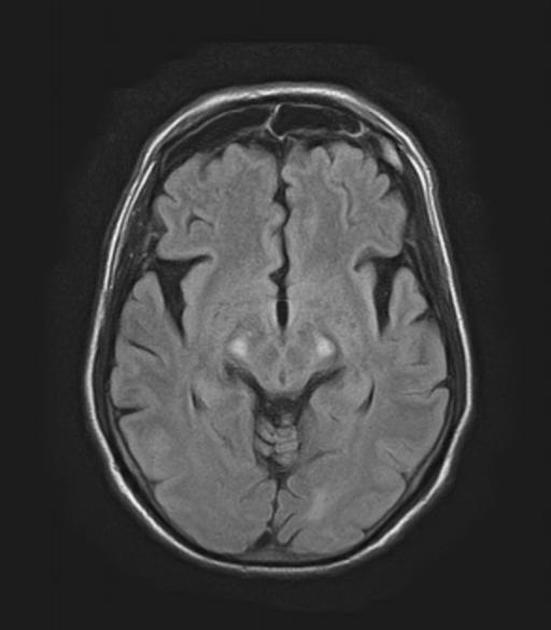
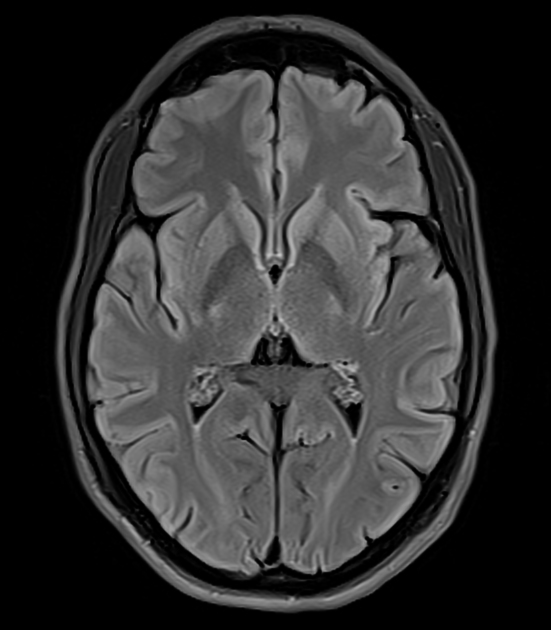
GRE/SWI: hypointensity in the precentral gyrus bilaterally, known as the "motor band sign" 8,9,14
-
MR spectroscopy 2
decreased NAA
decreased glutamate
increased choline
increased myo-inositol
Treatment and prognosis
Amyotrophic lateral sclerosis typically progresses to death in 2-6 years, usually from respiratory complications 5. Few therapies have been shown to extend survival, including riluzole, a glutamate antagonist 10,11. In patients with SOD1 mutations, the antisense oligonucleotide tofersen may be used 15.
History and etymology
Amyotrophic lateral sclerosis is also known as Charcot disease in honor of French neurologist Jean-Martin Charcot (1825-1893) who diagnosed and described the first case in the nineteenth century (1865-1869) 12. It is also popularly known as Lou Gehrig disease, especially in North America, in honor of legendary New York Yankees first baseman Henry Louis (Lou) Gehrig (1903-1941), who was diagnosed with the disease in 1939.
Differential diagnosis
metabolic diseases involving corticospinal tracts



 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.