
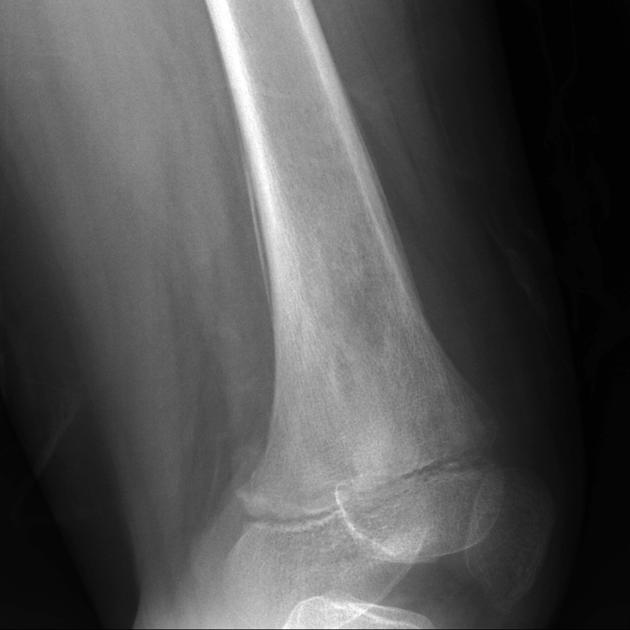
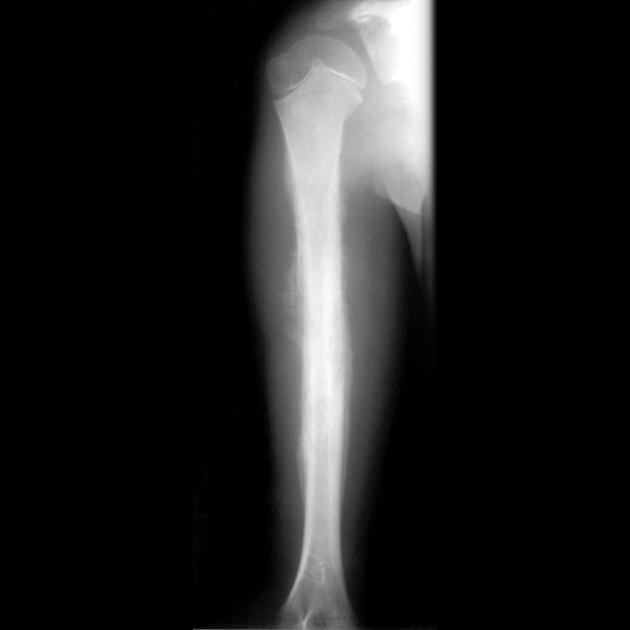
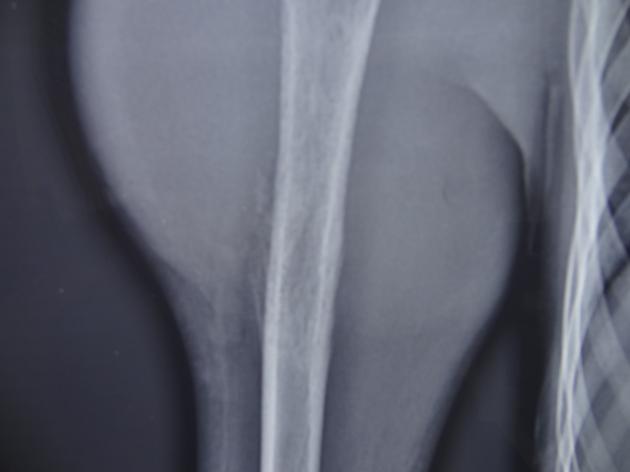
Ewing sarcomas are the second most common malignant primary bone tumors of childhood after osteosarcoma, typically arising from the medullary cavity with the invasion of the Haversian system. Ewing sarcomas usually present as moth-eaten, destructive, and permeative lucent lesions in the shaft of long bones, with a large soft tissue component and typical onion skin periostitis. These tumors may also involve flat bones and can appear sclerotic in up to 30% of cases.
On this page:
Epidemiology
Ewing sarcoma typically occurs in children and adolescents between 10-20 years of age (95% between 4-25 years of age) and has a slight male predilection (M:F 1.5:1) 1,2.
The Ewing sarcoma family of tumors primarily occurs in White patients. In the United States, the incidence in the Asian/Pacific Islander population is about one-half that in the White population, while the incidence in the Black population is one-ninth that in White population 12.
Clinical presentation
Presentation is non-specific with local pain being by far the most common symptom. Occasionally a soft tissue mass may be palpable. Pathological fractures also occur. Systemic symptoms including fever may be present. ESR and serum LDH levels are also elevated 14.
Pathology
Ewing sarcoma is a small round blue cell tumor with regular-sized primitive-appearing cells. It is closely related to the soft tissue tumors pPNET, Askin tumor, and neuroepithelioma, which collectively are referred to as Ewing sarcoma family of tumors (ESFT) 1. They share not only microscopic appearances but also demonstrate a non-random t(11;22)(q24;q12) chromosome rearrangement.
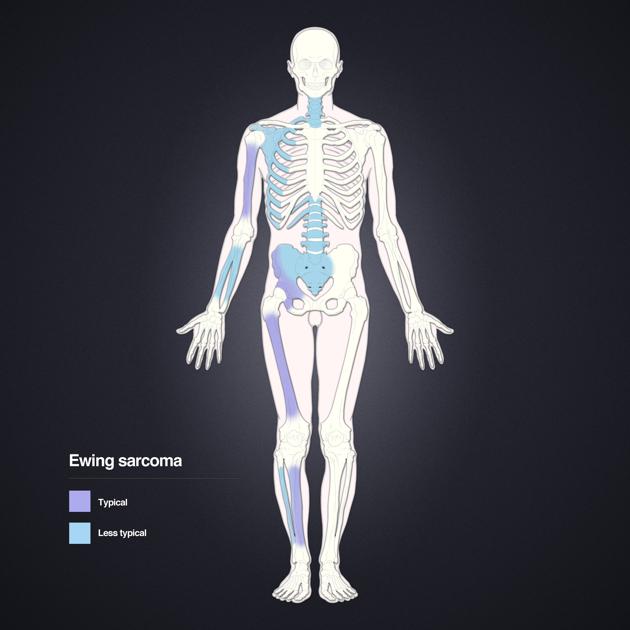
Location
-
lower limb: 45%
femur most common
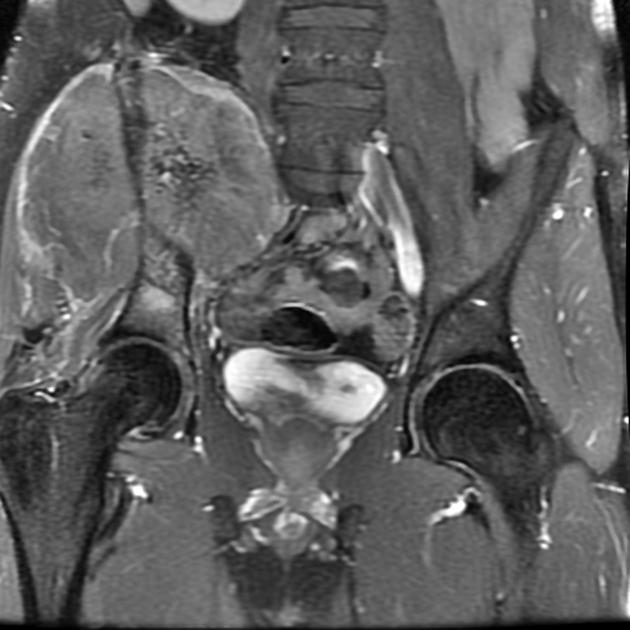
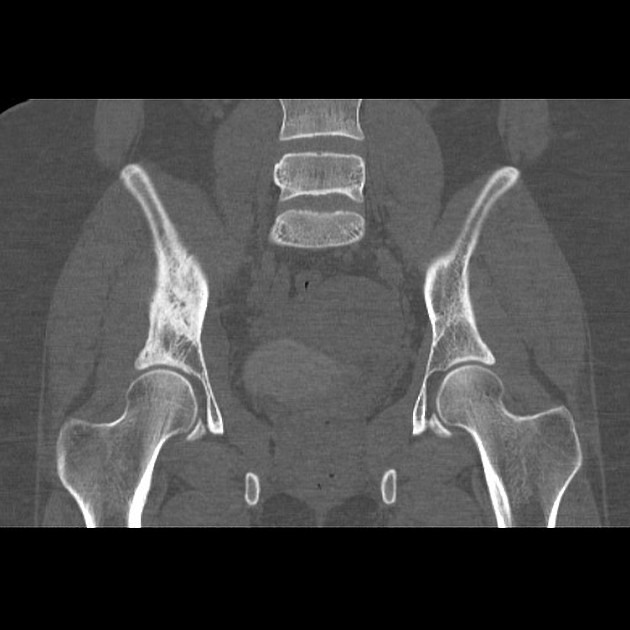
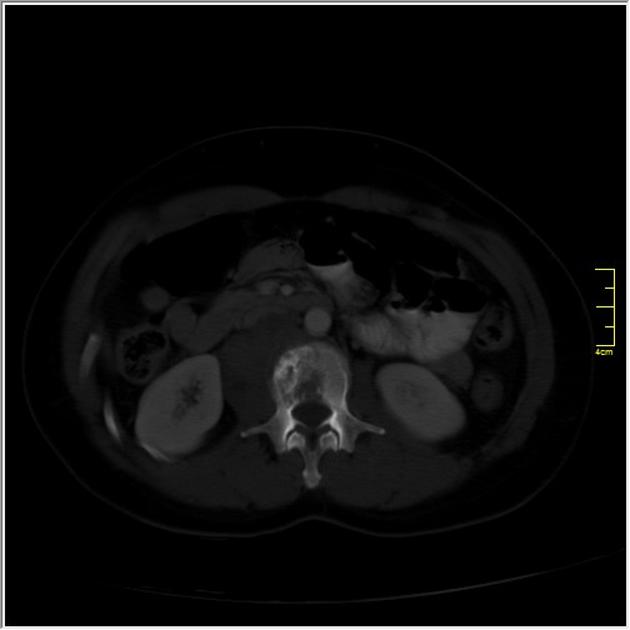
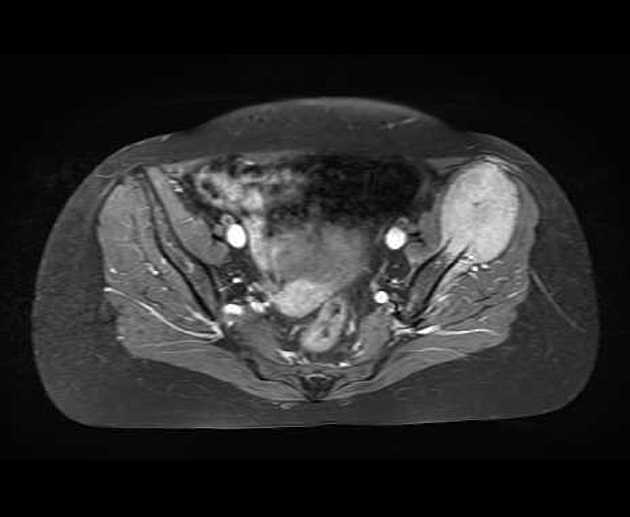
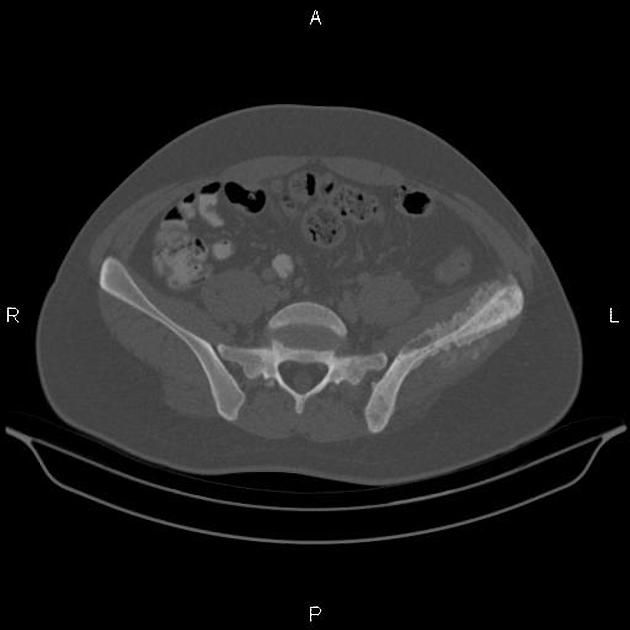
pelvis: 20%
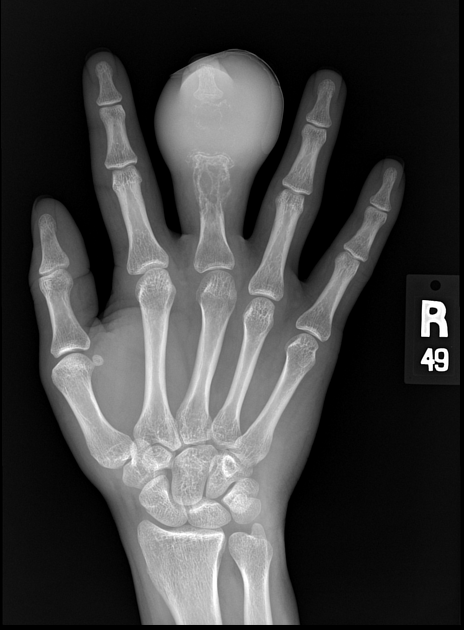
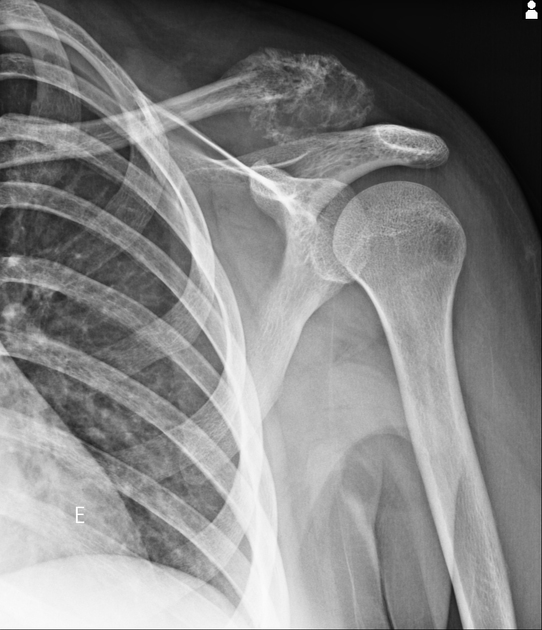
upper limb: 13%
-
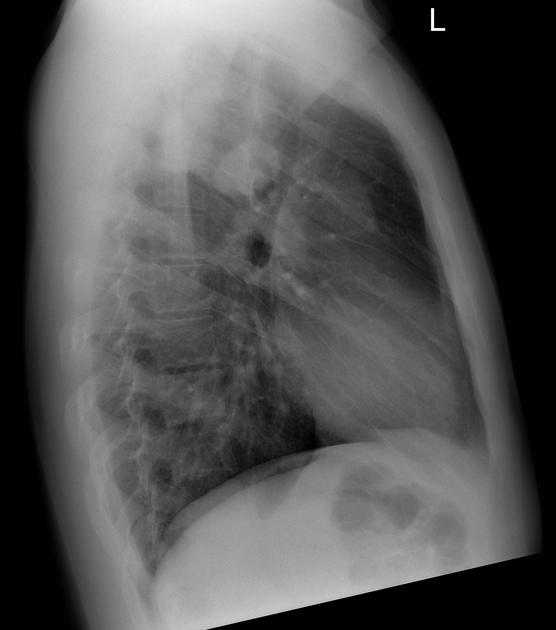
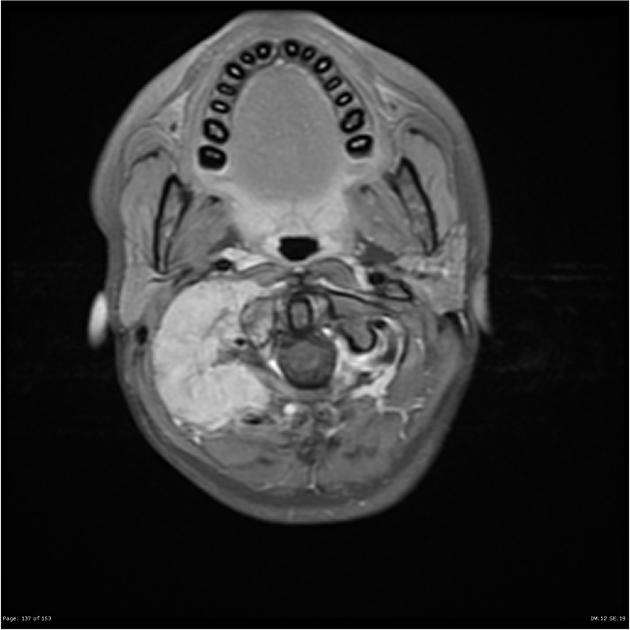
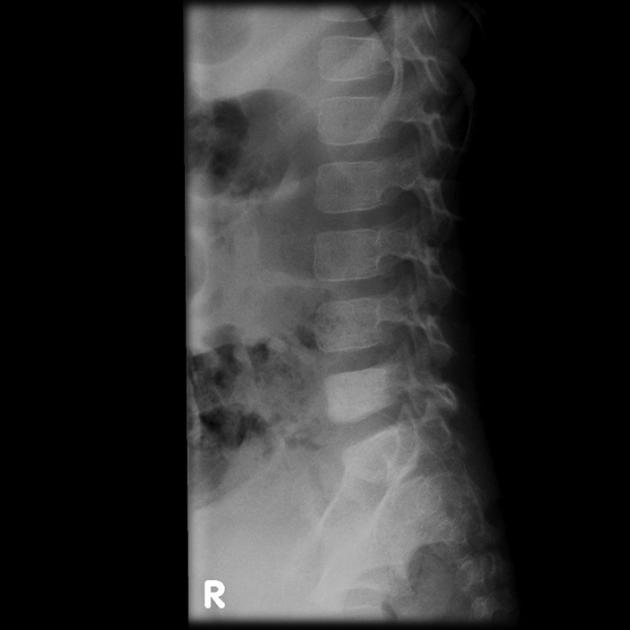
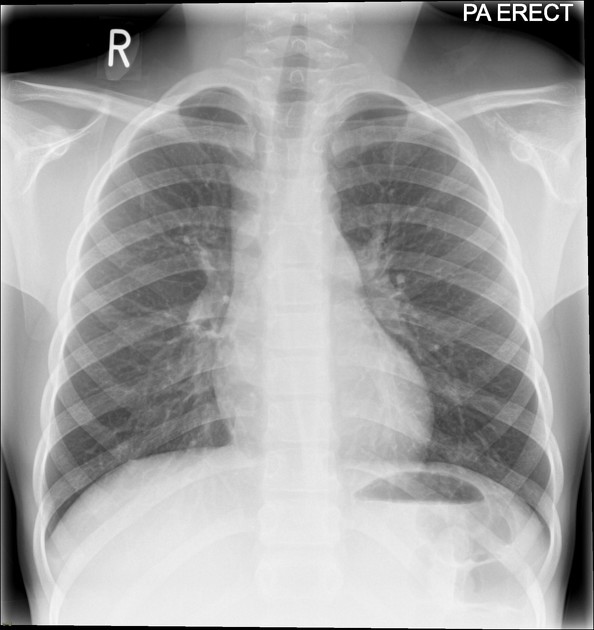
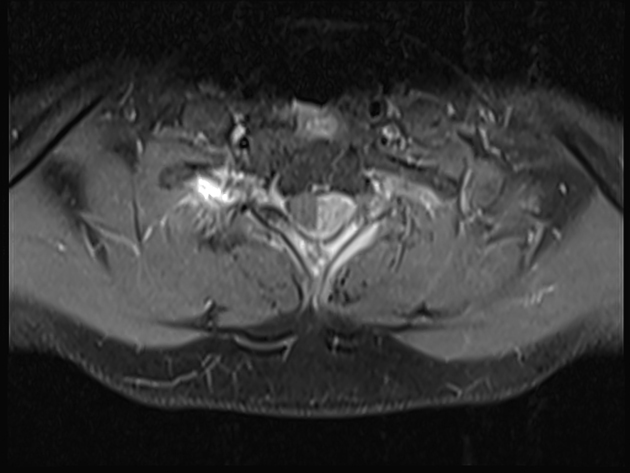
spine and ribs: 13% (thoracic Ewing sarcoma)
sacrococcygeal region most common 4
skull/face: 2%
Alternatively 3:
-
long bones: 50-60%
femur: 25%
tibia: 11%
humerus: 10%
-
flat bones: 40%
pelvis: 14%
scapula
ribs: 6% (thoracic Ewing sarcoma)
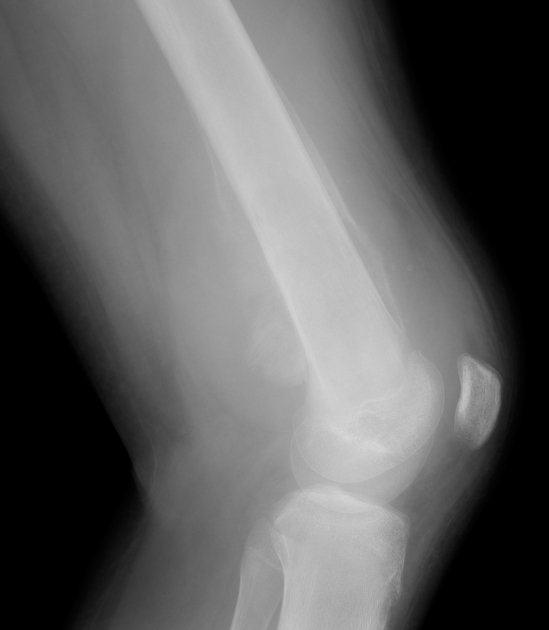
As far as a location within long bones, the tumor is almost always metadiaphyseal or diaphyseal 2,3:
mid-diaphysis: 33%
metadiaphysis: 44%
metaphysis: 15%
epiphysis: 1-2%
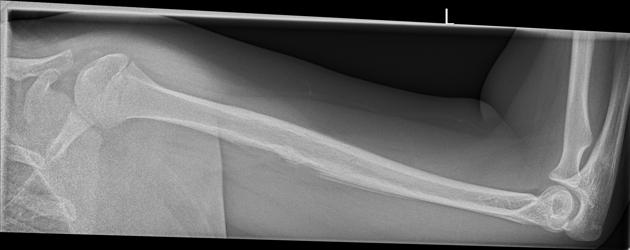
Radiographic features
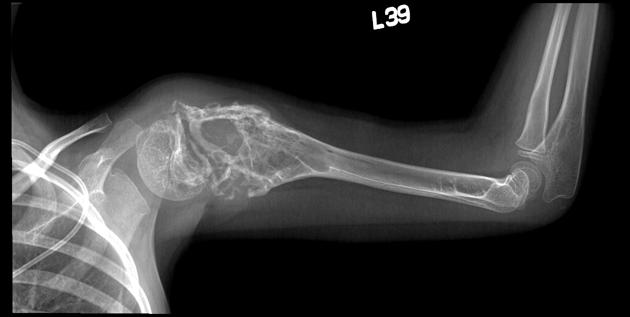
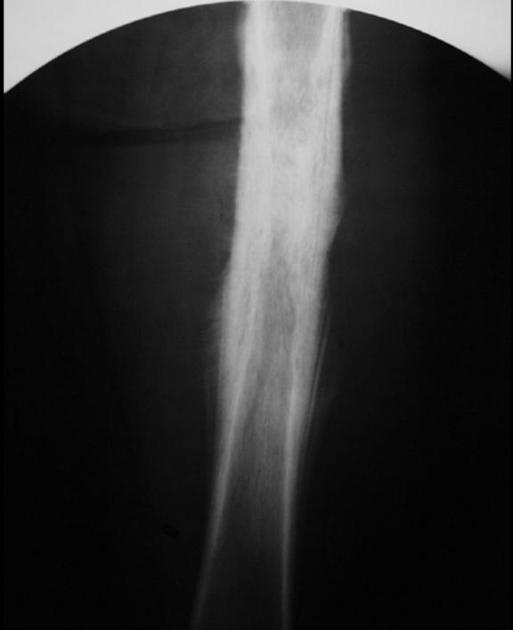
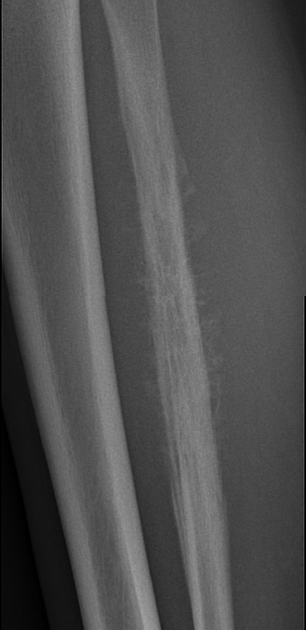
Ewing sarcomas tend to be large with a wide zone of transition/poorly defined margin. Over 80% of Ewing sarcomas demonstrate extension into adjacent soft tissues. It should be noted that pPNET often extend into bone, making the distinction difficult.
Plain radiograph / CT
The appearance of these tumors is very variable, but they usually have a clearly aggressive appearance. Common findings include 2,14:
permeative: 76%
sclerosis: 40%
They occasionally demonstrate other appearances, including Codman triangles, spiculated (sunburst) or thick periosteal reaction, and even bone expansion or cystic components.
Soft tissue calcification is uncommon, seen in less than 10% of cases 2.
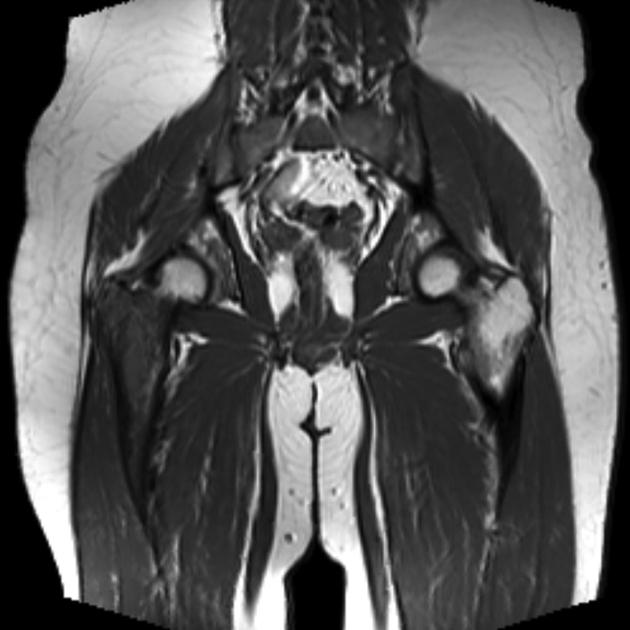
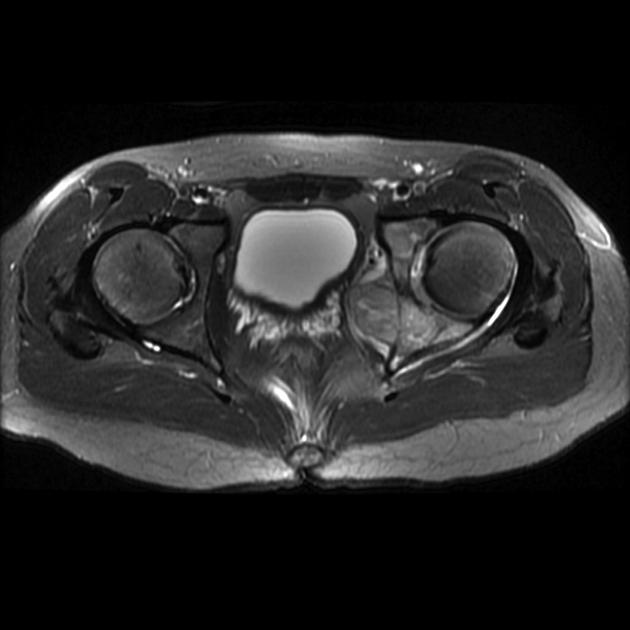
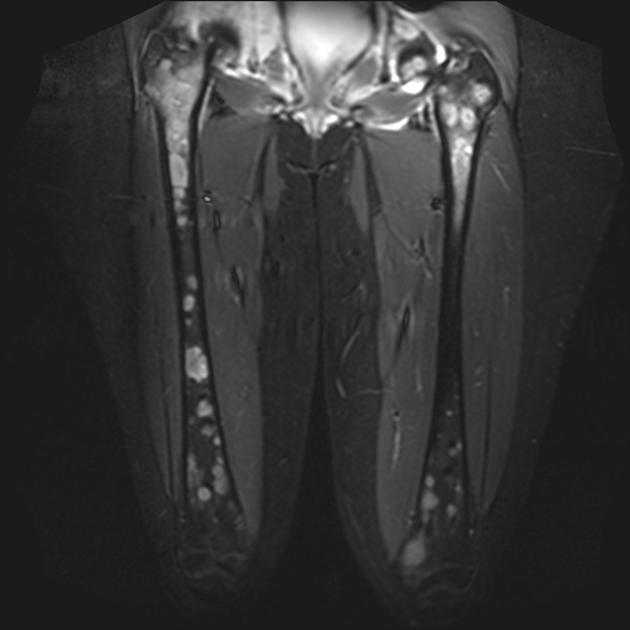
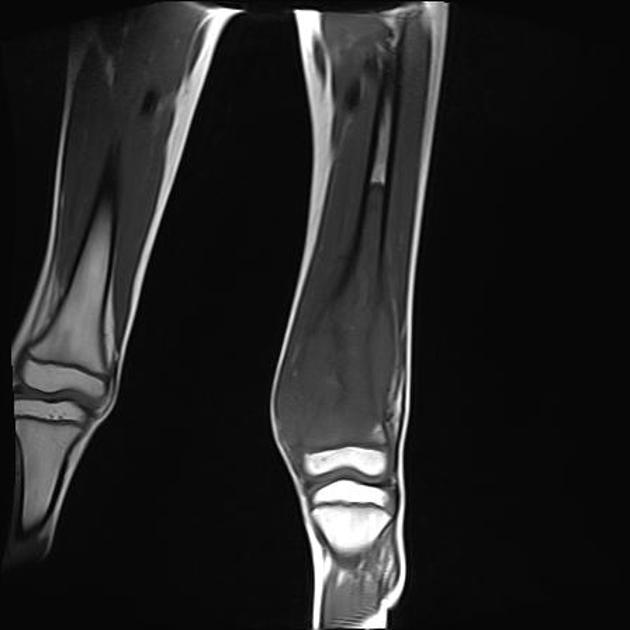
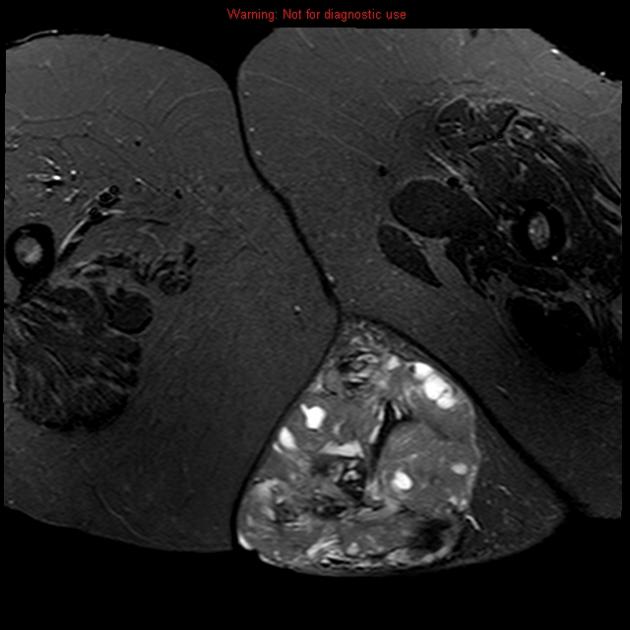
MRI
T1: low to intermediate signal
T1 C+ (Gd): heterogeneous but prominent enhancement
T2: heterogeneously high signal, may see hair on end low signal striations
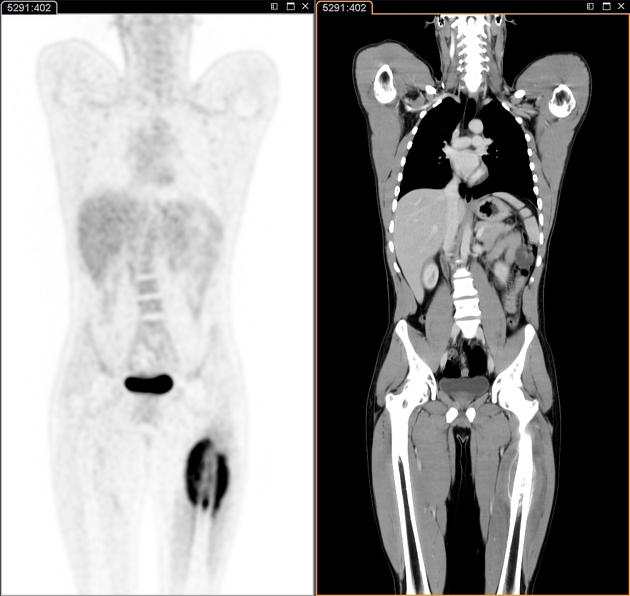
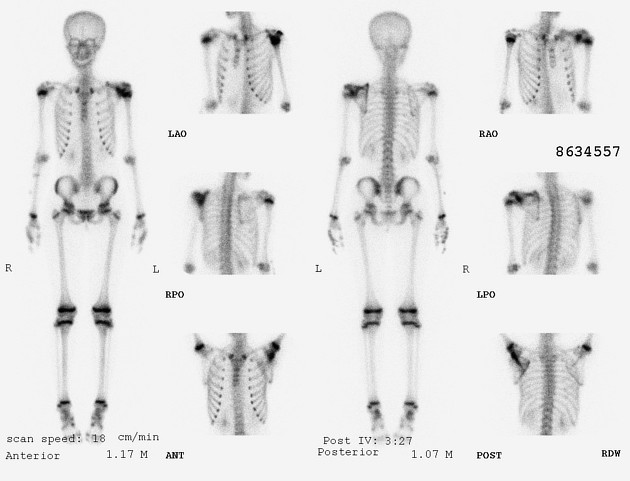
Nuclear medicine
Ewing sarcomas demonstrate increased uptake on both Gallium67 citrate and all three phases of Technetium99m methylene diphosphonate bone scans 6.
Treatment and prognosis
Systemic chemotherapy is the mainstay of treatment with surgery and/or radiotherapy playing a role depending on the location and size of the tumor.
What was once a uniformly fatal tumor now has respectable survival rates, although these vary with location. Spinal tumors for example have up to 86% long-term survival compared to 25% of sacrococcygeal tumors 4. The overall 5-year survival is in the order of 50-75% of patients with local disease only at the time of presentation 5.
Prognosis is significantly impacted by the presence of distant metastases at the time of diagnosis, which is far more common for the pelvis (25-30%) compared to extremities (<10%) 5.
Metastases most frequently go to lungs and bones in equal proportions. The spine is the most common site of bone metastases 13.
History and etymology
The tumor is named after James Stephen Ewing (1866-1943), an American pathologist, who first described his eponymous tumor in 1920 8,11.
Differential diagnosis
-
other Ewing sarcoma family of tumors
pPNET: large soft tissue component with extension into bone
Askin tumor: chest wall
-
more often has amorphous calcified matrix
classically perimetaphyseal, Ewing sarcoma also occurs in other locations
more prevalent around the knee and in the proximal humerus, in other locations Ewing sarcoma is the more frequent of the two
metastatic disease
hematological malignancy





 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.