
Autosomal recessive polycystic kidney disease (ARPKD) is one of many pediatric cystic renal diseases.
On imaging, it usually presents on ultrasound with enlarged echogenic kidneys with multiple small cysts. Liver involvement with coarse echotexture, biliary tract cystic changes, and portal hypertension may be evident, depending on the age and stage of hepatic fibrosis.
On this page:
Epidemiology
ARPKD is one of the commonest inheritable infantile cystic renal diseases but is far less common than autosomal dominant polycystic kidney disease (ADPKD) which affects adults. The incidence is estimated at ~1:20,000-50,000 6,9. There is no perceived gender or racial predilection.
Associations
congenital hepatic fibrosis: the degree of which is inversely proportional to the age of presentation 3,4
Clinical presentation
The age of presentation is variable and is divided into perinatal, neonatal, infantile, and juvenile forms (for the sake of simplicity one can think of only two groups: those that present around birth, and those that present later in childhood). There is an inverse relationship with the severity of associated congenital hepatic fibrosis. In patients who present early (perinatal/neonatal) renal disease dominates the clinical picture, whereas in older children (infantile/juvenile) liver disease dominates 9.
Pathology
Results from a mutation in the PKHD1 (polycystic kidney and hepatic disease) gene location on chromosome 6p. This results in bilateral symmetric microcystic disease occurring in the distal convoluted tubules and collecting ducts. The number of ducts involved determines the age of presentation.
-
perinatal type: most common
75% have death within 24 hours of delivery
minimal hepatic fibrosis
neonatal type: minimal hepatic fibrosis
infantile type: moderate periportal fibrosis
-
juvenile type: gross hepatic fibrosis
Radiographic features
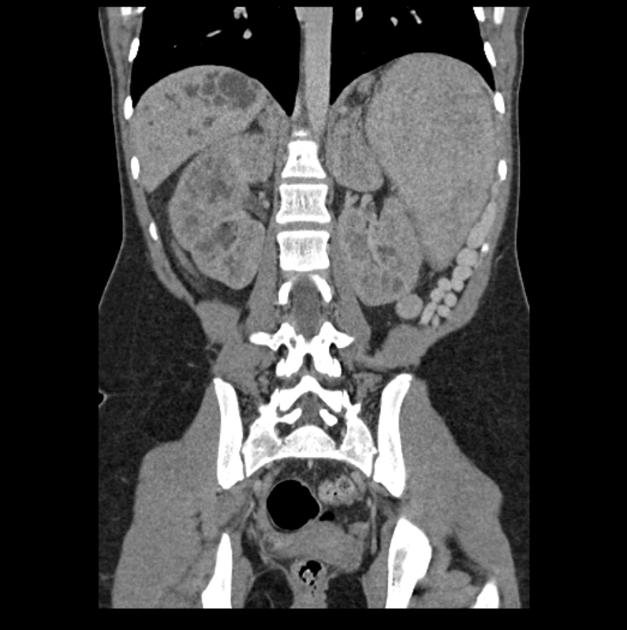
Involvement is usually bilateral, and often first assessed with ultrasound.
Fluoroscopy
Intravenous pyelography (IVP)
If performed, a streaky appearance may be demonstrated, representing the ectatic collecting ducts.
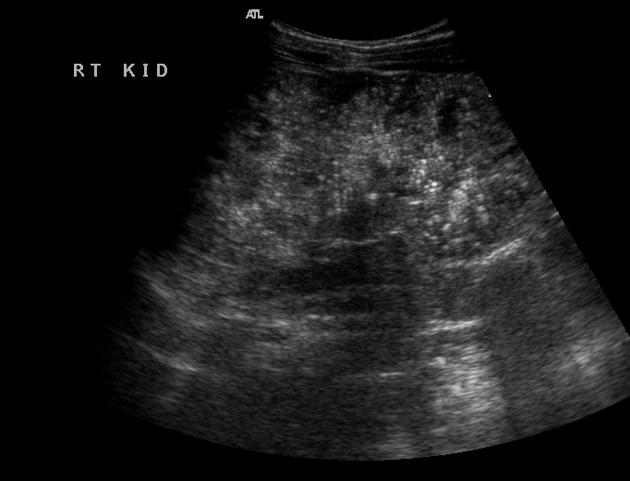
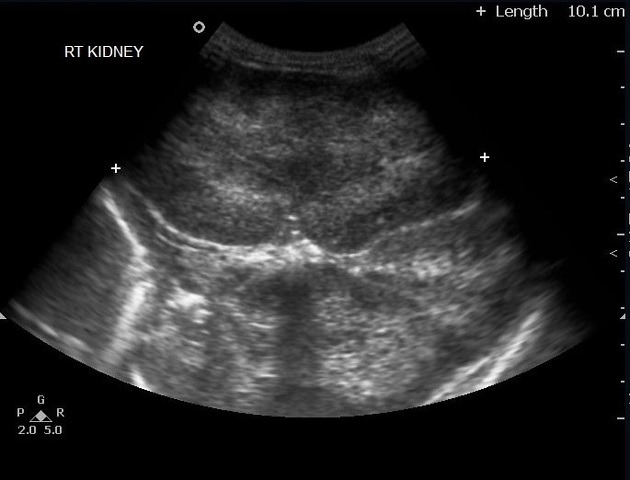
Ultrasound
Appearances somewhat depend on the age of the patient 8,9:
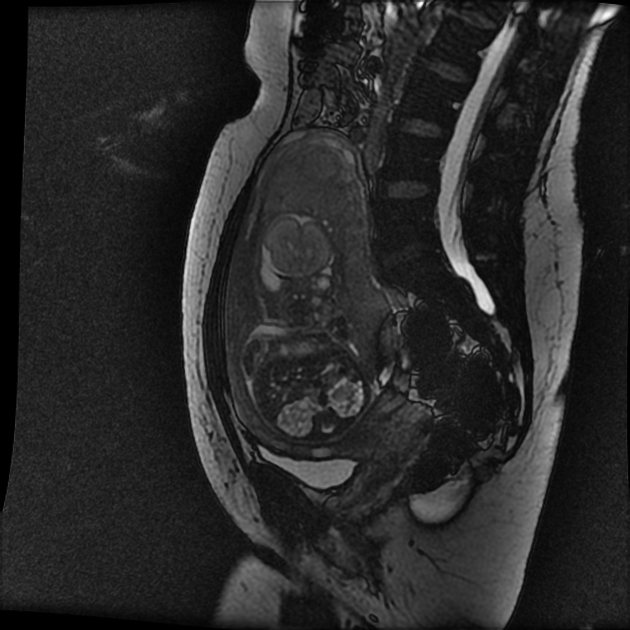
on antenatal ultrasound, associated oligohydramnios may be identified
-
cysts
initially too small to resolve but with time may become discernible
unlike ADPKD, the cysts rarely exceed 1-2 cm in diameter
the kidneys appear enlarged and echogenic but usually, retain a reniform shape
-
medullary pyramids
initially may appear hypoechoic compared to the cortex: which can give a peripheral halo during this stage
eventually become increasingly hyperechoic
corticomedullary differentiation is eventually lost
high-resolution ultrasound (linear-array transducer, 7.5 MHz or greater) allows visualization of numerous cylindrical cysts in the medulla and cortex, which represent ectatic collecting ducts 8
Ultrasound is also useful in assessing the liver (see: congenital hepatic fibrosis) and may demonstrate 9:
normal or coarsened echotexture
biliary tract cystic change (Caroli disease)
portal hypertension and sequelae thereof
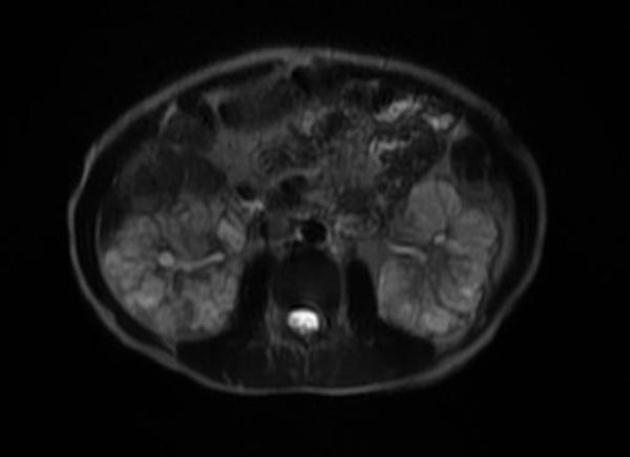
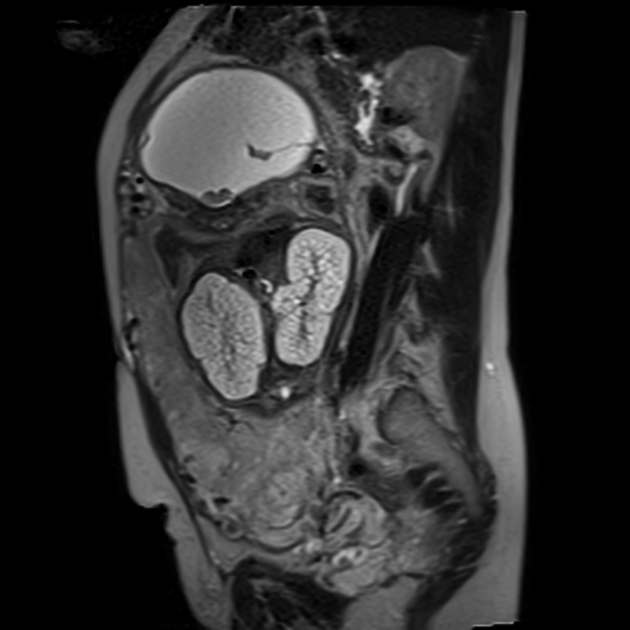
MRI
frequently shows enlarged kidneys with diffusely increased T2 signal
may also show evidence of oligohydramnios in better detail
Treatment and prognosis
Treatment is largely supportive. Dialysis and transplantation (renal +/- liver) may be offered.
The prognosis is poor in general but also significantly depends on the type. Overall, ~40% (range 30-50%) of affected infants die during the perinatal period owing to pulmonary hypoplasia and pulmonary insufficiency 8.
Complications
Differential diagnosis
For enlarged echogenic kidneys on an antenatal ultrasound scan, considerations include:
autosomal dominant polycystic kidney disease (ADPKD): the large cysts may not form in utero, and the kidneys may initially appear as enlarged and echogenic
renal dysplasia associated with trisomy 13








 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.