
Autosomal dominant polycystic kidney disease (ADPKD), also sometimes referred to as "adult polycystic kidney disease", is an inherited form of adult cystic renal disease.
On this page:
Epidemiology
Autosomal dominant polycystic kidney disease is one of the most common serious hereditary diseases, found in 1:400 to 1:1000 individuals, and is by far the most common inherited cause of end-stage renal failure (ESRF) 6. It accounts for 4-10% of all cases of ESRF 6.
Associations
Several conditions are well recognized as being associated with ADPKD 1,3,5,6:
-
found in 6% of patients with ADPKD without a family history of aneurysms
found in up to 22% of patients with ADPKD with a family history 16
intracranial dolichoectasia: 2-3% 6
hypertension: up to 80% of adults 6
small bowel diverticula (perhaps) 5
mitral valve prolapse: up to 25% 3
multiple biliary hamartomas (von Meyenberg complexes) 9
-
cysts in other organs
liver: most common, 75% by age 60 years 6
spleen: ~5 %
seminal vesicles: 60% by age 40 years 6
prostate: 11%
-
pancreas: ~10%:
pancreatic cysts are more common in von Hippel Lindau disease (vHL)
Diagnosis
Genetic testing is costly, so diagnosis is commonly made by a combination of family history and cyst detection. Advances in technology mean that smaller cysts can be identified and this has led to suggestions to modify the criteria, e.g. two or more cysts in each kidney in US in an at-risk individual aged 30-40 years17. High-resolution T2 FSE is highly sensitive for subcentimeter cysts and more objective. Ten or more cysts in the 16-49 year age group were found to have sensitivity and positive predictive value of 100% in at-risk individuals; a limit of five cysts can be used to identify suitable related renal donors 17.
Clinical presentation
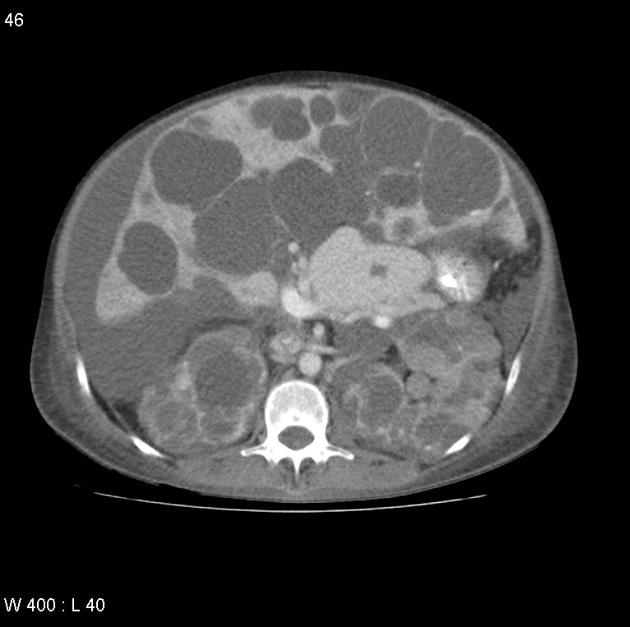
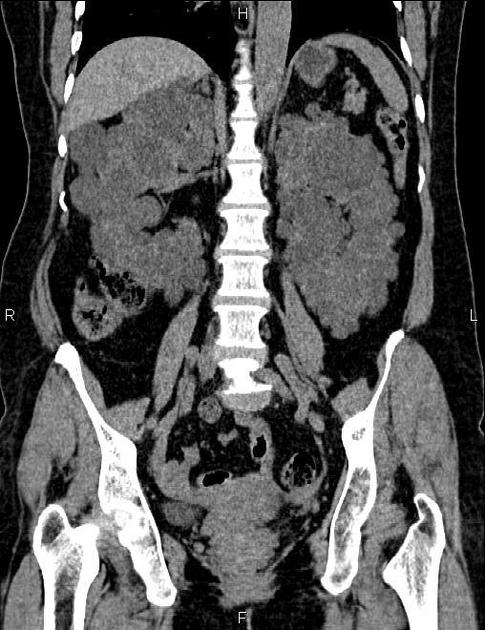
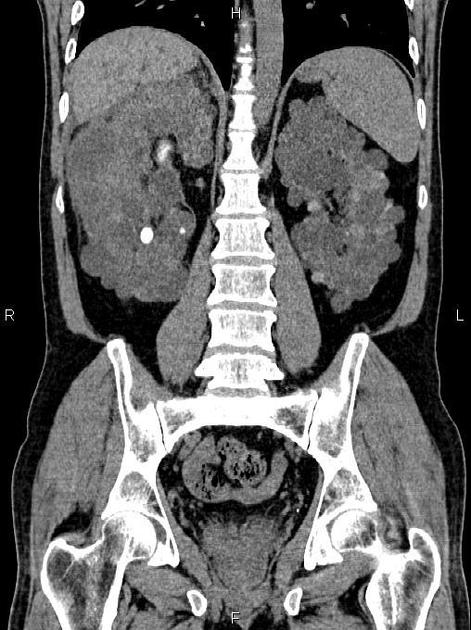
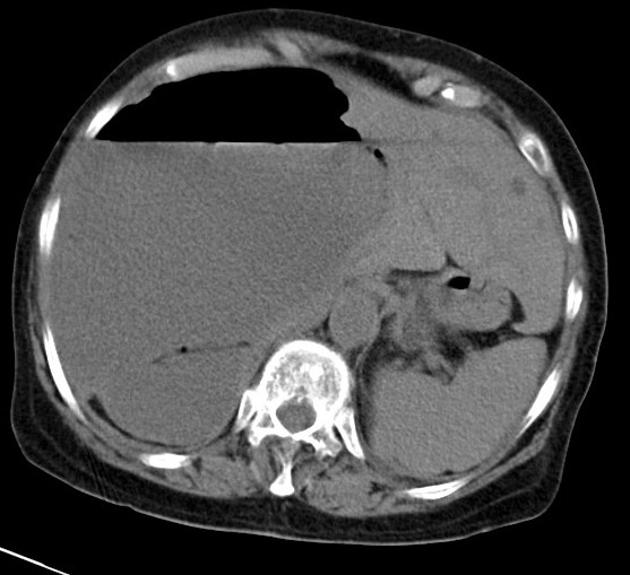
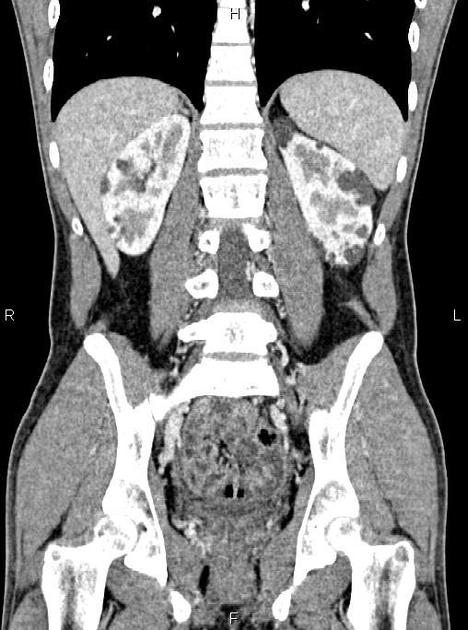
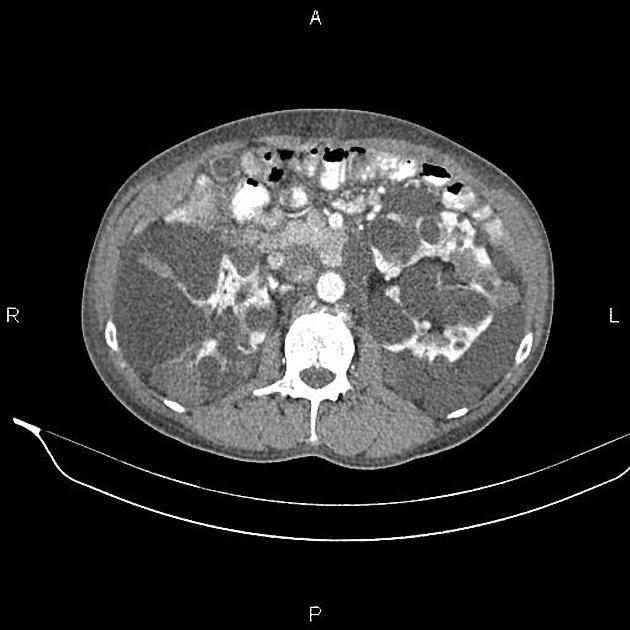
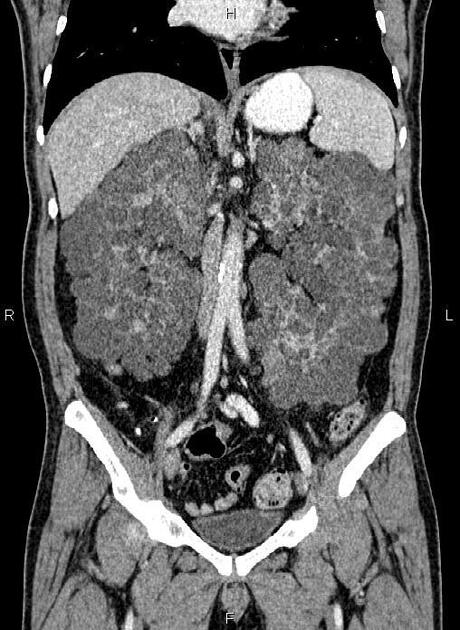
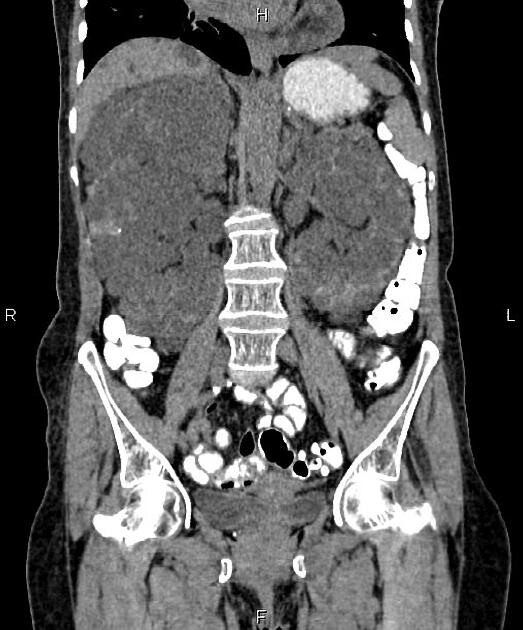
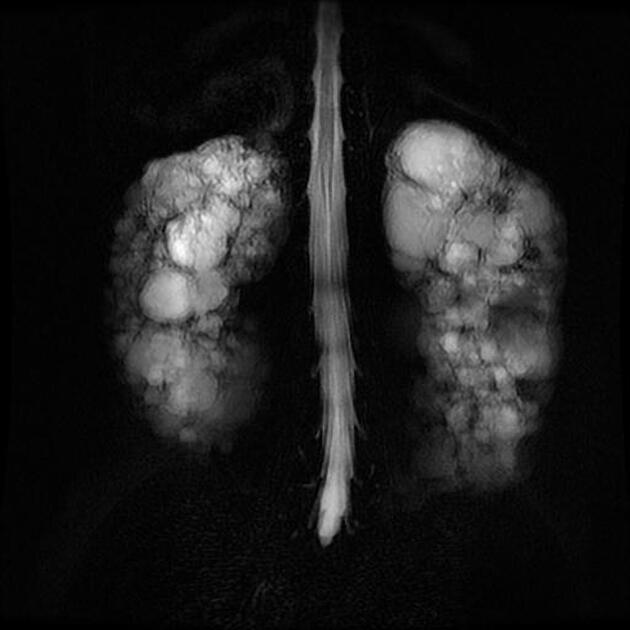
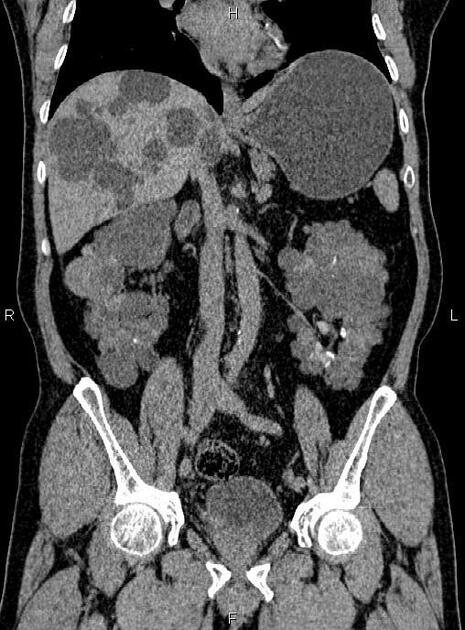
The kidneys are normal at birth, and with time develop multiple cysts. At the age of 30 years, approximately 68% of patients will have visible cysts by ultrasound 3. Cyst number and size increase with age and all patients eventually demonstrate cystic change. By the age of 60 years, approximately 50% of patients have end-stage renal failure. The risk of renal cancer is not increased.
Clinical presentation is variable and includes 1:
dull flank pain of variable severity and time course: most common
abdominal or flank masses
hematuria
hypertension: usually develops at the same time as renal failure
renal functional impairment to renal failure
Pathology
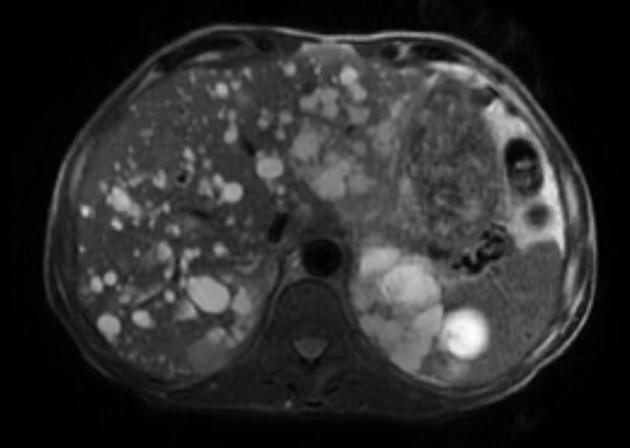
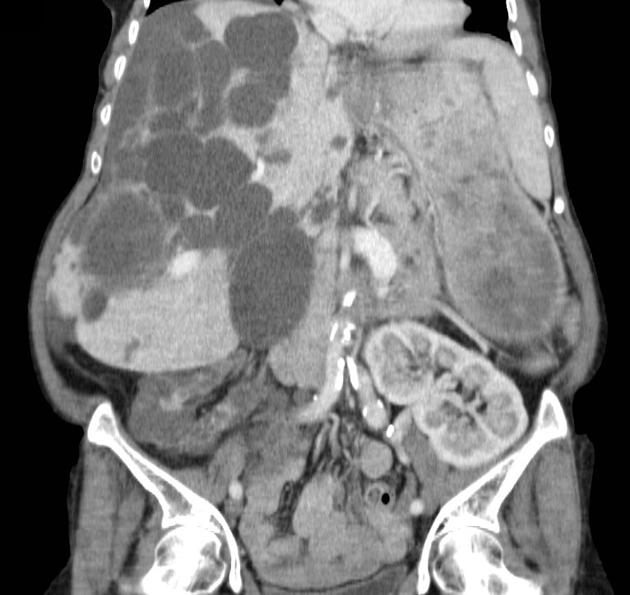
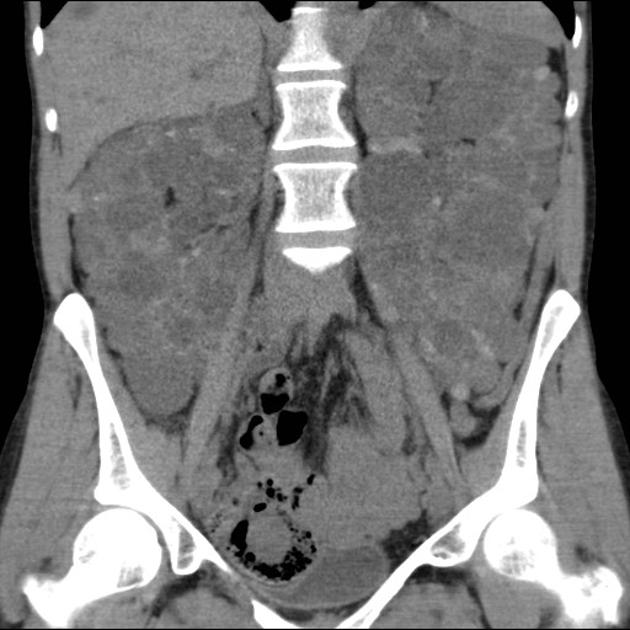
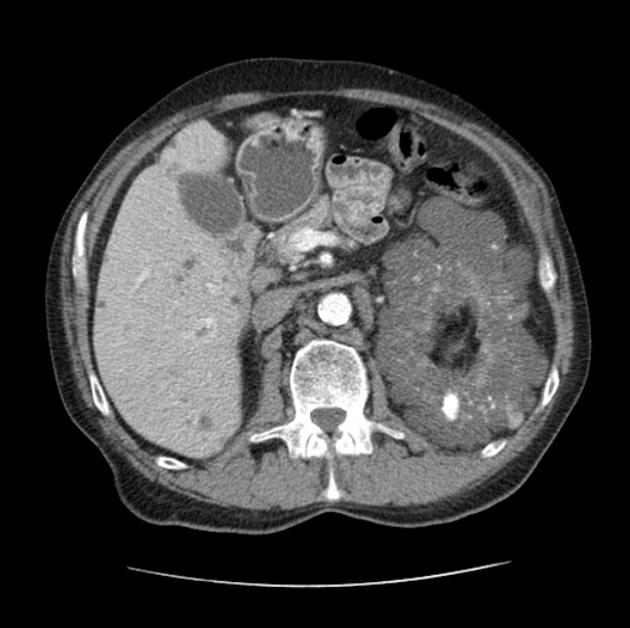
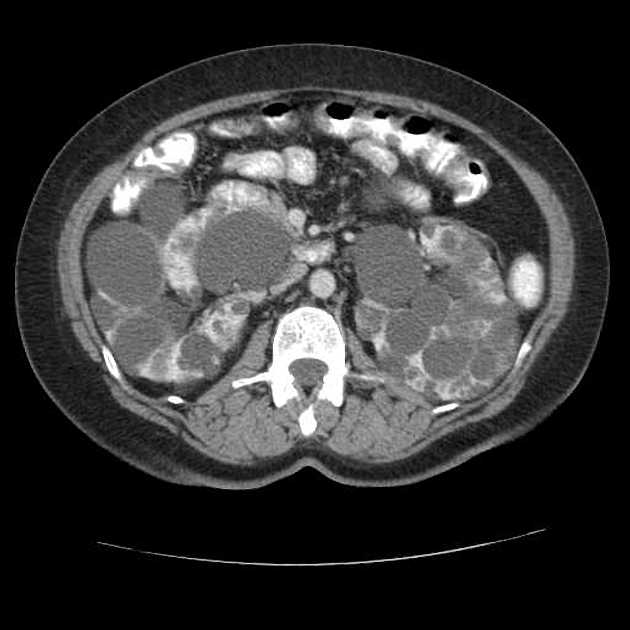
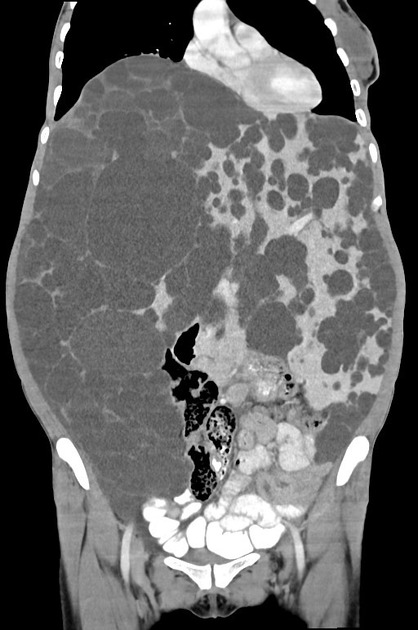
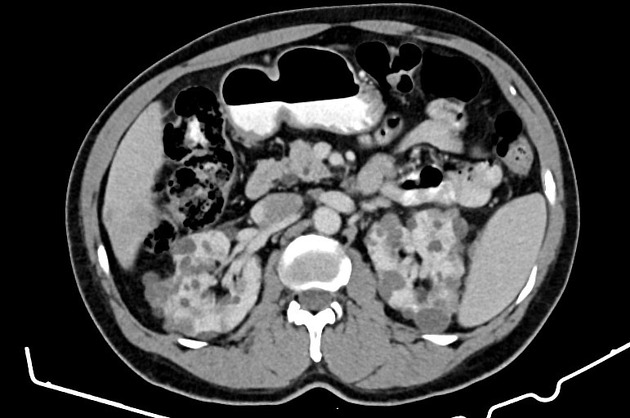
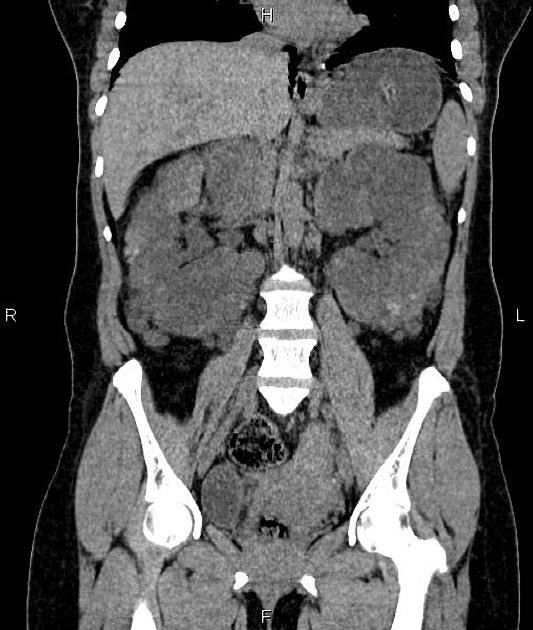
Macroscopically the kidneys demonstrate a large number of cysts of variable size (from a few mm to many cm), in both the cortex and medulla. They are filled with fluid of variable color (from clear or straw-colored to altered blood or chocolate-colored to purulent when infected).
Genetics
The majority of cases are inherited in an autosomal dominant fashion. In around 28% of cases, no family history is apparent16 and the disease may be due to a spontaneous mutation 1.
Three genes have been identified, with slightly different phenotypes 1,3,13:
-
PKD1
located on chromosome 16p
80% of cases
encodes polycystin-1 transmembrane protein
presentation is earlier and more likely to progress to end-stage renal failure, average age of 54 years16
-
PKD2
chromosome 4q
15% of cases
encodes polycystin-2 14
less severe, ESRD at an average age 74 years16
-
GANAB (rare)
chromosome 11q13
liver cysts common +/- hepatic dysfunction
mild renal disease, ESRF unusual
The main abnormality is in the cilia-centromere complex of tubular epithelial cells 14. The defect results in cystic dilatation of the renal tubules (of all parts of the nephron) in a minority of nephrons. The cysts are variable in size, resulting in compression and atrophy of the remainder of the kidney, increased renin and erythropoietin secretion, and gradual renal dysfunction.
Radiographic features
Full imaging assessment of patients with autosomal dominant polycystic kidney disease can be challenging, simply due to the size and number of the cysts and associated mass effect on adjacent structures. All cysts need to be assessed for atypical features, that may reflect complications (e.g. bleeding or infection) or malignancy (i.e. renal cell carcinoma) 2.
Enlargement of the kidneys is associated with poorer renal function. Therefore, renal volume measurement can be used to predict the risk of chronic renal failure 15.
Plain radiograph
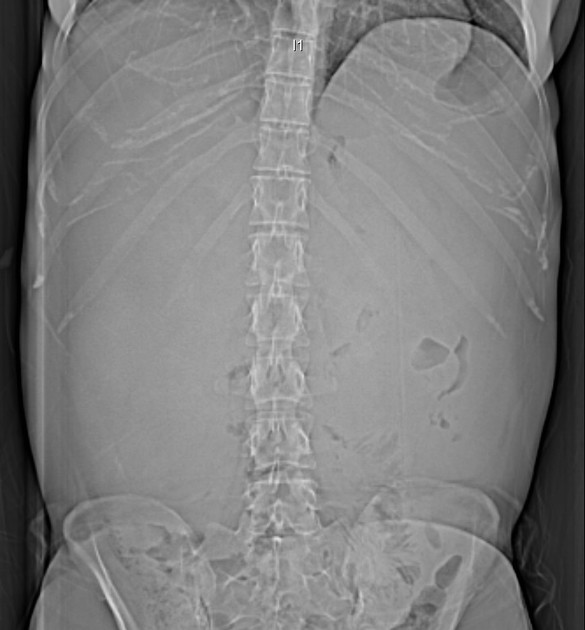
Plain films have no role in the surveillance of patients with established ADPKD. The diagnosis may be suspected when the renal outlines are enlarged, multilobulated or difficult to discern, with associated displacement of loops of bowel.
Multiple calcifications may be seen which may have multiple ring configurations.
On intravenous urography, "Swiss cheese nephrogram" is seen due to multiple radiolucencies noted due to multiple renal cysts. "Spiderleg pyelogram" is also described since stretched out and attenuated pelvicalyceal system is seen as a result of the mass effect caused by renal cysts.
Ultrasound
Ultrasound is fast, relatively inexpensive and lacks ionizing radiation. It can suggest the diagnosis and assess for cyst complications.
Simple renal cysts will appear anechoic with well-defined thin walls, posterior acoustic enhancement (amplification) and lateral shadowing (extinction) 3.
Cysts with hemorrhage or infection will demonstrate echogenic material within the cyst, without internal blood flow. Calcification may develop in cyst walls. Renal cell carcinomas in contrast, although usually cystic in the setting of ADPKD, will have solid components with blood flow. CEUS is useful to demonstrate enhancement.
Perinephric and intrarenal hematomas may occur in the context of minor trauma.
Cysts may be detected in the liver, spleen, pancreas and seminal vesicles.
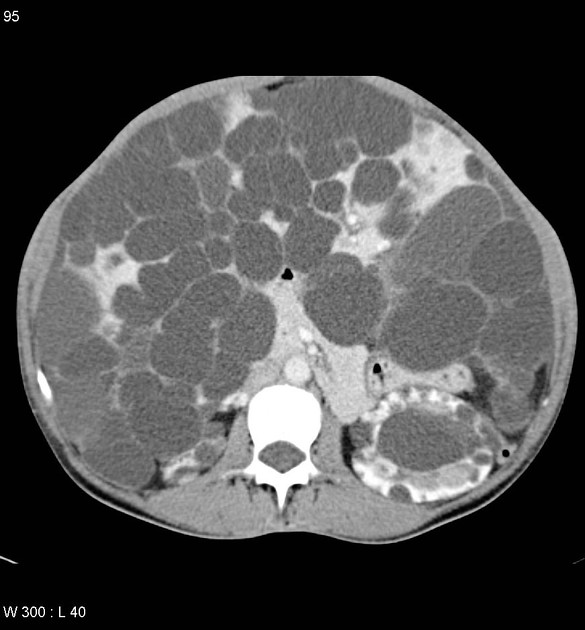
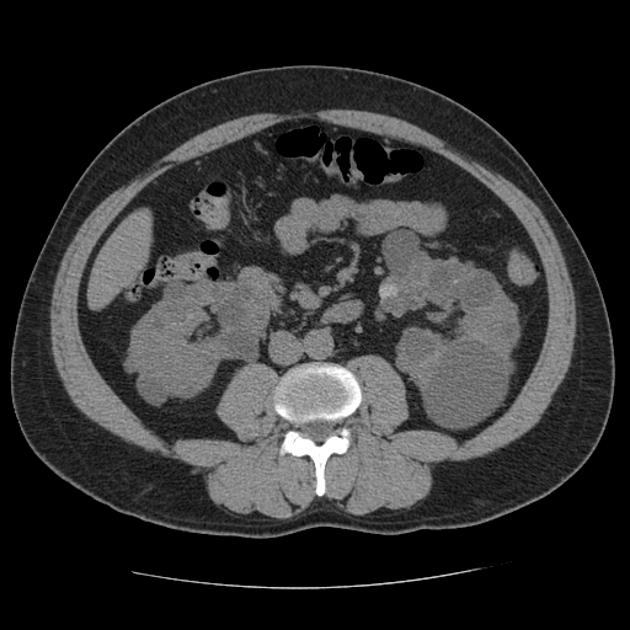
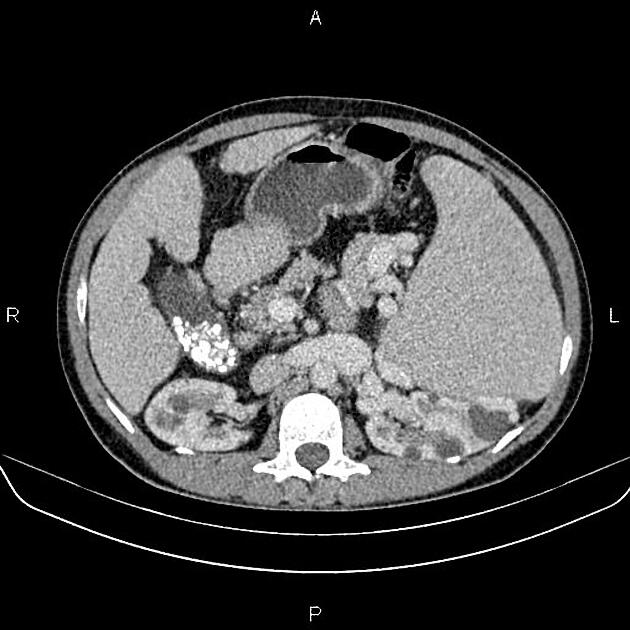
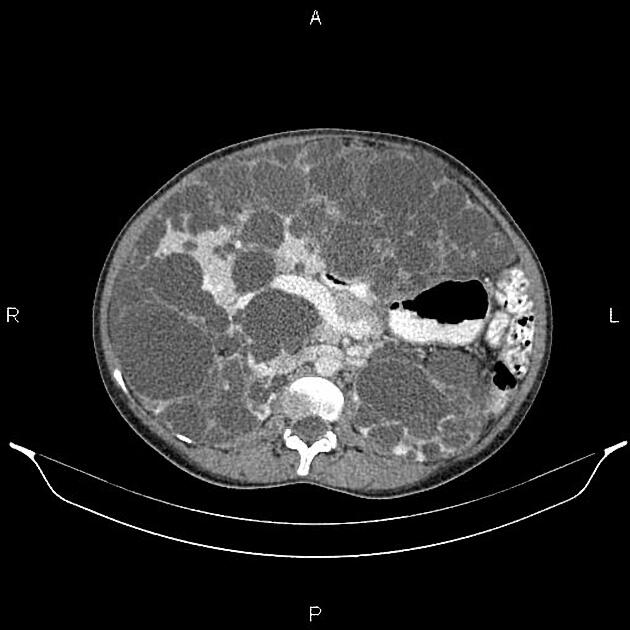
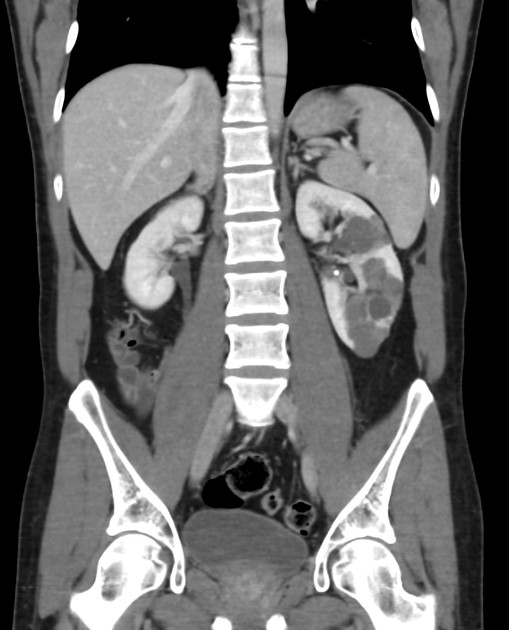
CT
Simple cysts appear as rounded structures with near-water attenuation and thin, regular walls. Complications such as hemorrhage or infection raise the attenuation of the cyst contents and induce dystrophic wall calcification.
A complex cystic mass with an enhancing solid component may indicate renal cell carcinoma (RCC). The Bosniak classification of renal cysts is of limited value in ADPKD. Subtraction CT, MRI or DECT are sensitive to enhancement.
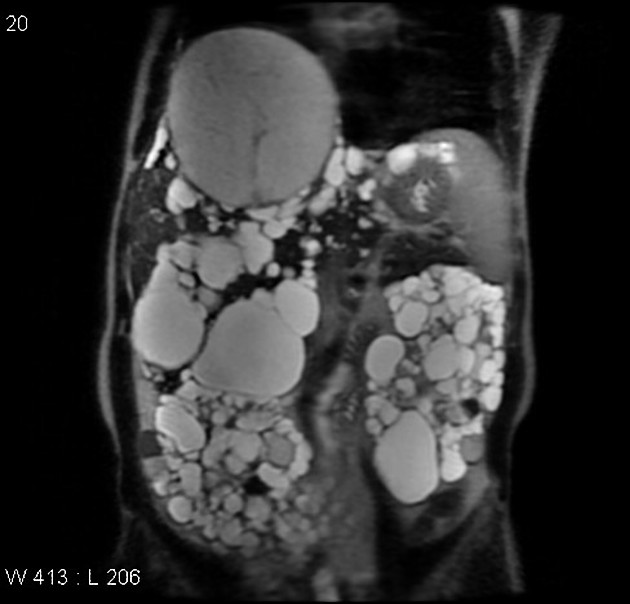
MRI
High-resolution T2-weighted MRI is the most sensitive modality for the detection of small renal cysts. Simple cysts are thin-walled and the contents appear similar to water.
T1: low signal
T2: high signal
-
T1 C+ (Gd)
consider using ACR group II ultra-low-risk contrast media to avoid nephrogenic systemic fibrosis
simple cysts should not have any solid-enhancing components
presence of enhancement of a solid component or septa should raise the possibility of a renal cell carcinoma (RCC) (N.B. infected cysts may peripherally enhance, as do islands of trapped renal tissue). Subtraction MR may be helpful.
Treatment and prognosis
ESRF requiring transplant or dialysis eventually develops in many patients (45% by the age of 60). Patients with PKD1 mutations are more likely to progress to ESRF and often do so at an earlier age 1. Patients with a larger height-adjusted total kidney volume are at greater risk of ESRF and may benefit from vasopressin receptor antagonists which slow cyst expansion16. EGFR is often stable until the disease is advanced, therefore size and morphology are more useful predictors of outcome. The Mayo Clinic Imaging Classification uses size and morphology to predict disease course. Radiomics and textural analysis show promise in predicting decline.
Complications
Complications may be both local (i.e. of the kidney) or of other organ systems.
Renal complications include 1,3:
progression to end-stage renal failure
recurrent urinary tract infections
cyst hemorrhage or infection resulting in acute pain
cyst rupture: resulting in retroperitoneal hemorrhage
calculi: urinary stasis promotes stone formation
Unlike in some other congenital cystic kidney diseases, there is no increased risk for renal cell carcinoma (RCC) unless the patient is undergoing prolonged dialysis 11.
Distant complications include:
Differential diagnosis
General imaging differential considerations include:
-
von Hippel Lindau disease (vHL)
renal cysts and RCCs
pancreatic cysts and tumors
pheochromocytoma
contiguous gene syndrome: large deletions of chromosome 16 with overlapping features of APKD and TSC
-
primary renal disease-related cysts
the renal parenchyma appears abnormal, reduced in volume with increased echogenicity on ultrasound 3
-
multiple 'incidental' renal cysts
usually far fewer in number
kidney not enlarged
hydronephrosis mimicking multiple cysts
acquired cystic kidney disease: occurs in those with chronic renal failure (particularly in those on dialysis)
-
autosomal recessive polycystic kidney disease (ARPKD)
enlarged kidney
cysts are very numerous and small
changes are present in childhood
corticomedullary differentiation is lost
-
cysts smaller and located in the medulla cortex junction 7
-
may be unilateral, unlike ADPKD
Practical points
diagnosis is usually made by imaging at-risk individuals
the height-adjusted total kidney volume for both kidneys and the quantity of remaining normal renal tissue are the best indicators of future renal function and the need for vasopressin receptor antagonists
















 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.