
Usual interstitial pneumonia (UIP) is not a disease, it is a histopathologic and radiologic pattern of interstitial lung disease. This can be caused by idiopathic pulmonary fibrosis, fibrotic hypersensitivity pneumonitis or non-specific interstitial pneumonia. The diagnosis is typically decided by a multidisciplinary team.
On this page:
Terminology
The term pneumonia derives from the pathological classification and does not imply an infective etiology.
Pathology
The histological diagnosis of UIP is based on temporal and spatial heterogeneity, the identification of fibrotic lesions at different stages (fibroblastic infiltrates, mature fibrosis, and honeycombing) within the same biopsy specimen and architectural distortion.
Honeycombing, particularly if it involves more than 5% of the lung volume, is an almost 100% specific finding. On a typical biopsy, there are areas of normal lung alternating with interstitial fibrosis and honeycombing. The distribution of UIP characteristically is with an apicobasal gradient with basal and peripheral (subpleural) predominance, although it is often patchy.
Inflammation is absent or mild and mostly limited to the areas of honeycombing 1-12.
Etiology
UIP pattern of interstitial lung disease can be seen in idiopathic pulmonary fibrosis or secondary to underlying systemic diseases. These would include:
-
connective tissue disorders (CTD associated UIP): falls under the broader spectrum of connective tissue disorder interstitial lung disease (CTD-ILD)
rheumatoid arthritis: UIP is considered to be the dominant pattern in those with rheumatoid arthritis who have concurrent interstitial lung disease 3
systemic sclerosis (scleroderma): either a UIP or nonspecific interstitial pneumonia (NSIP) (more common) pattern 4
polymyositis/dermatomyositis: a UIP, NSIP, or cryptogenic organizing pneumonia pattern 4
mixed connective tissue disease: either a UIP or NSIP pattern 4
asbestos-related interstitial lung disease: asbestosis 1
medications/drug toxicity: amiodarone lung
Hermansky-Pudlak syndrome (very rare)
In practice, the diagnosis is usually made in a multidisciplinary approach involving chest physicians, radiologists, and pathologists with expertise in interstitial lung disease 12.
Radiographic features
Plain radiograph
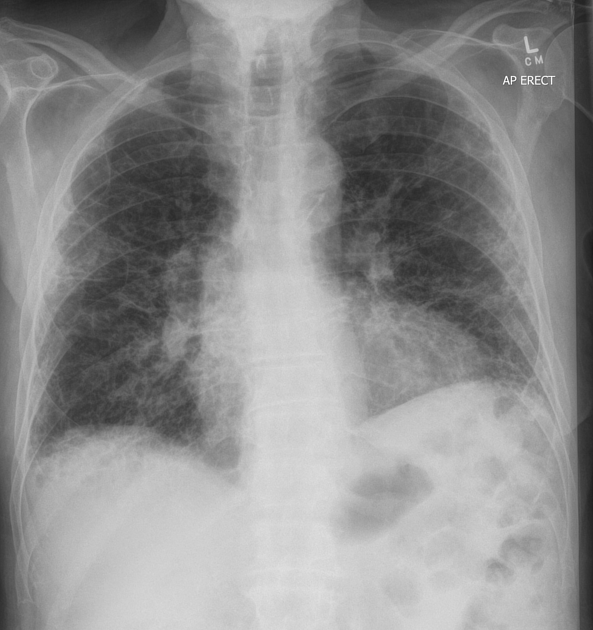
Plain film features are non-specific. While chest radiographs can be even normal in patients with very early disease, in advanced disease, it may show decreased lung volumes and basal fine to coarse reticulation. Usually, due to the more extensive involvement of the lower lobes, the major fissure is shifted inferiorly which is best seen on the lateral chest radiograph.
CT
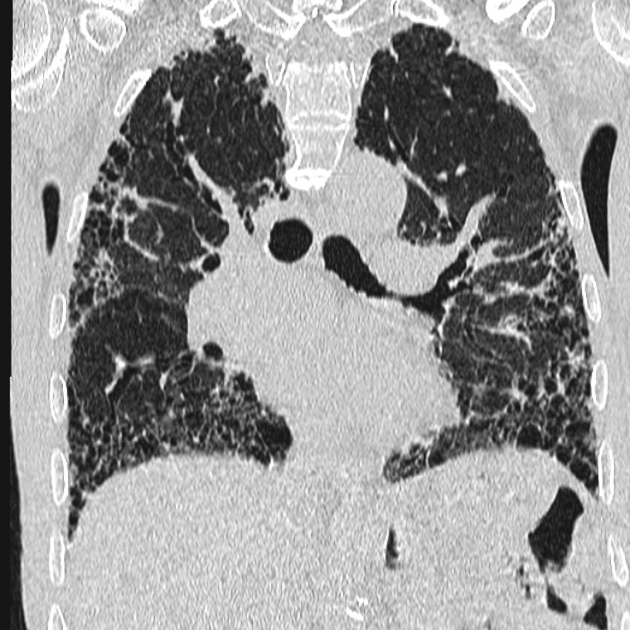
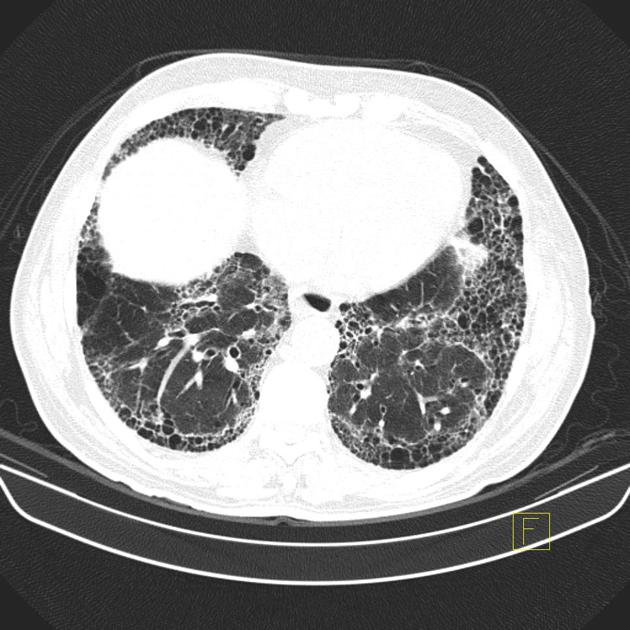
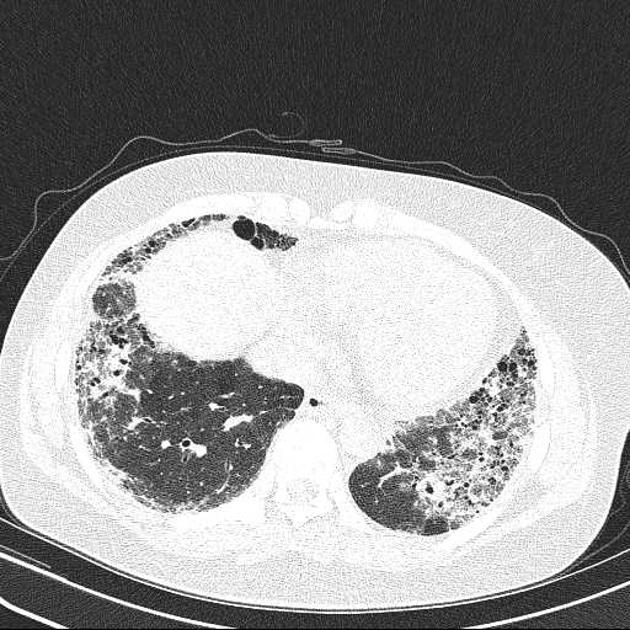
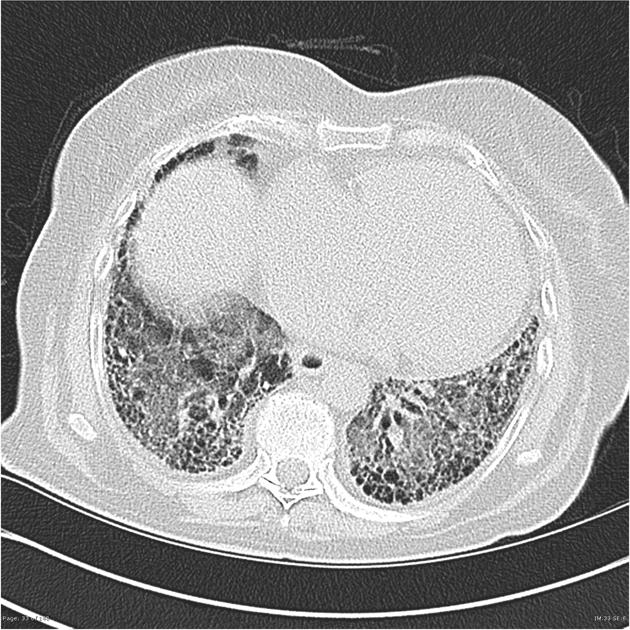
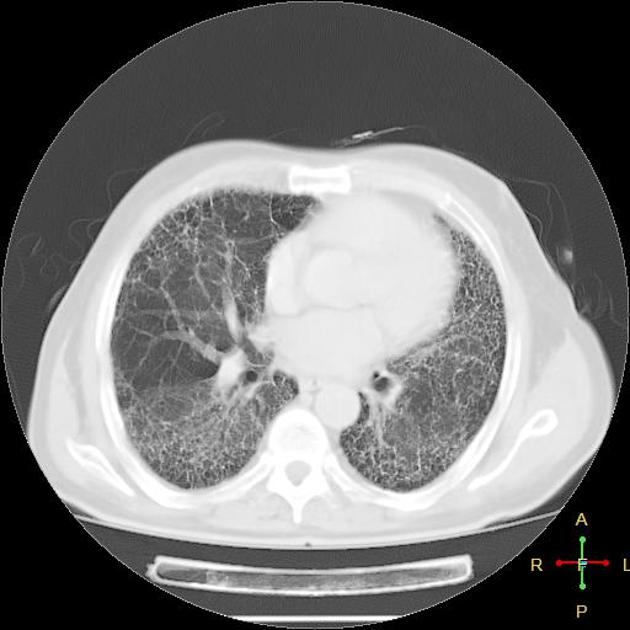
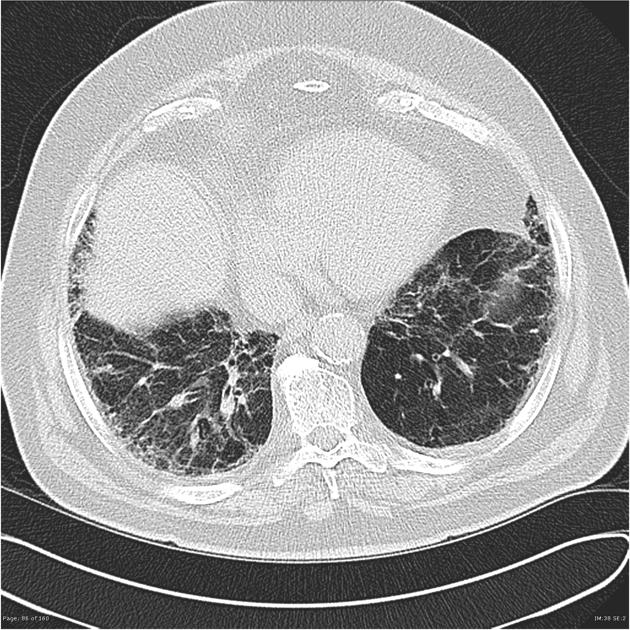
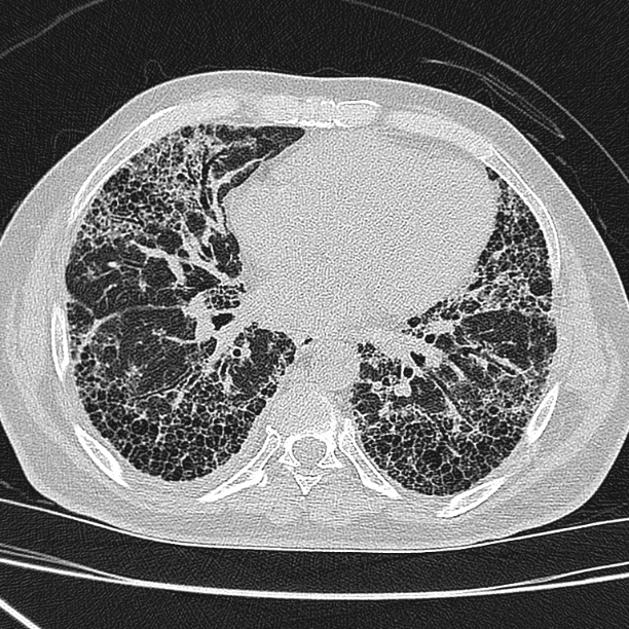
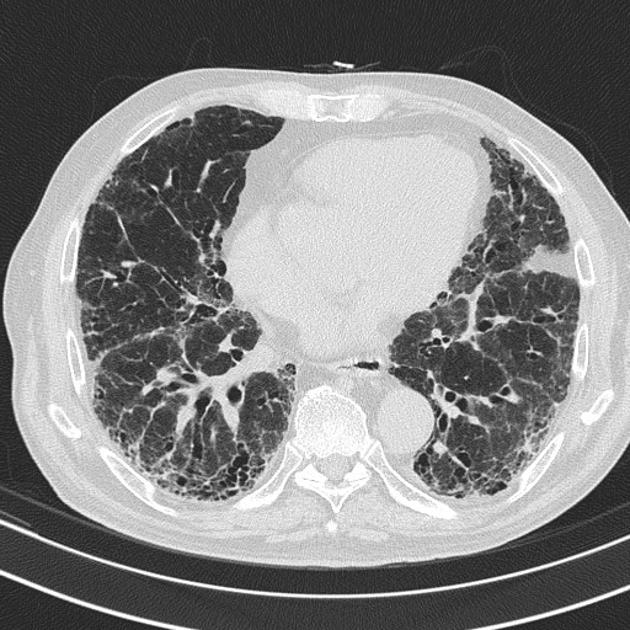
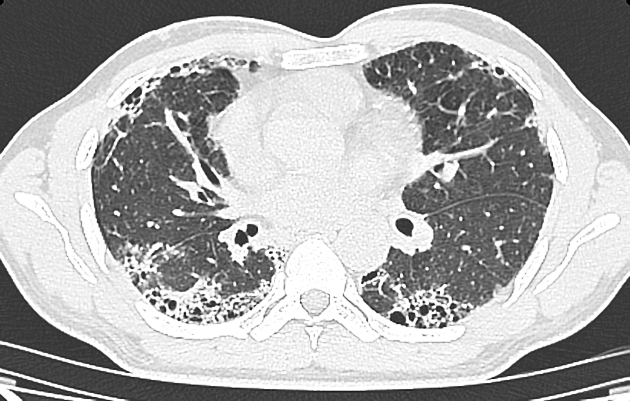
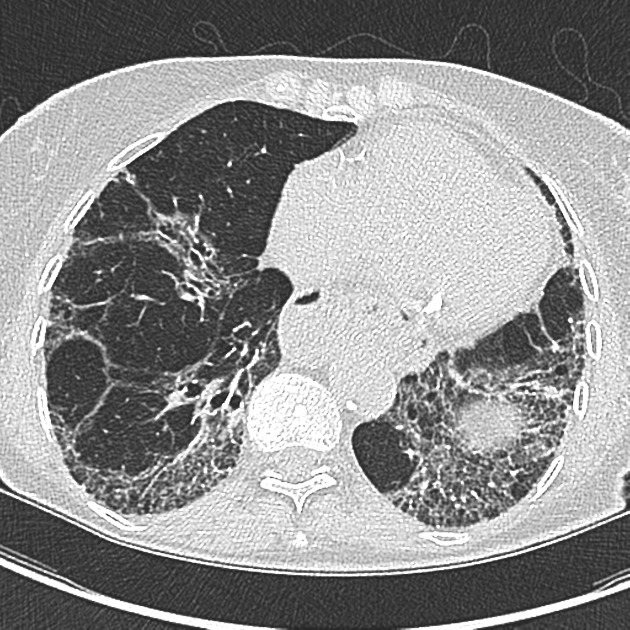
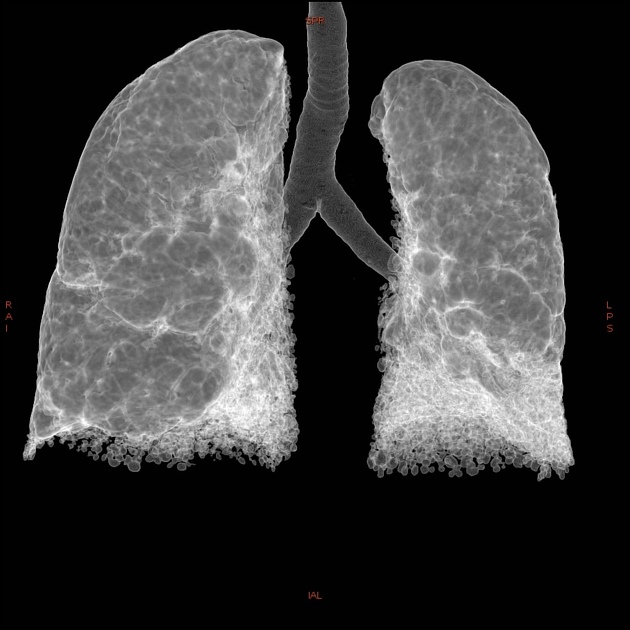
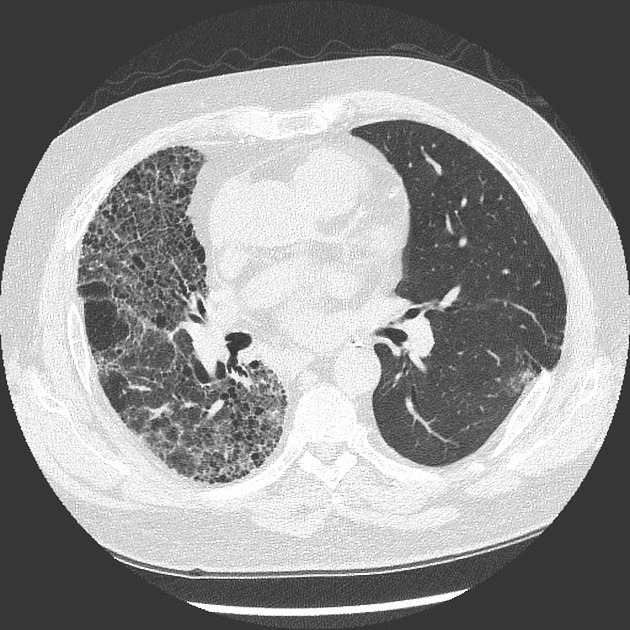
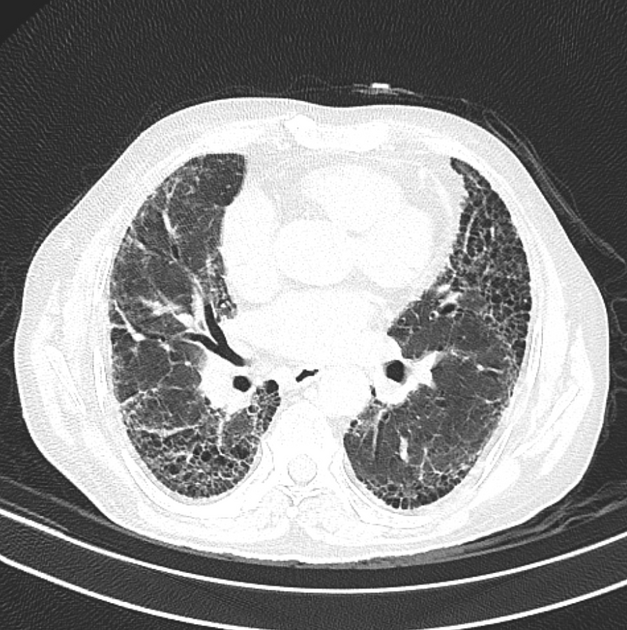
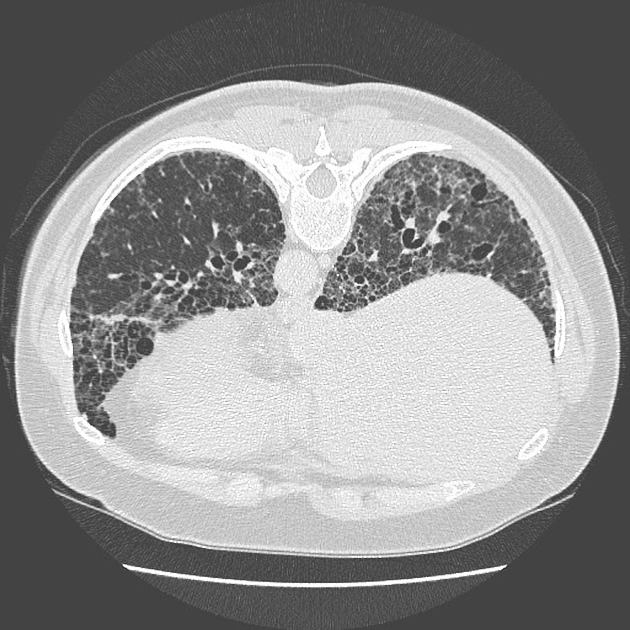
UIP pattern at CT has specific diagnostic criteria on HRCT 16 which include honeycombing, traction bronchiectasis and traction bronchiolectasis. The distribution is often widespread, asymmetric, and heterogeneous but with subpleural and basal predominance 23. Nodular pulmonary ossification may be seen in fibrotic areas.
The positive predictive value of CT in the diagnosis of UIP is high and ranges from 70 to 100% 1. Similar to the pathology specimen, cross-sectional imaging also reveals heterogeneity, with patchy areas of fibrosis alternating with areas of normal lung 5.
Typical features include 1,5:
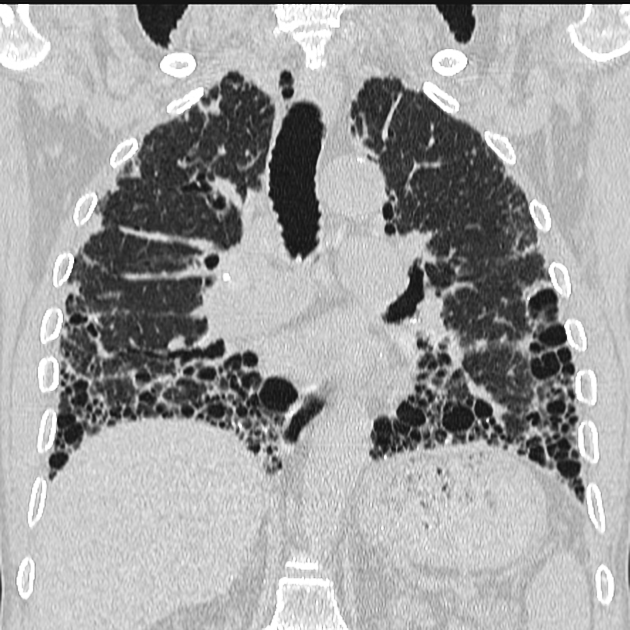
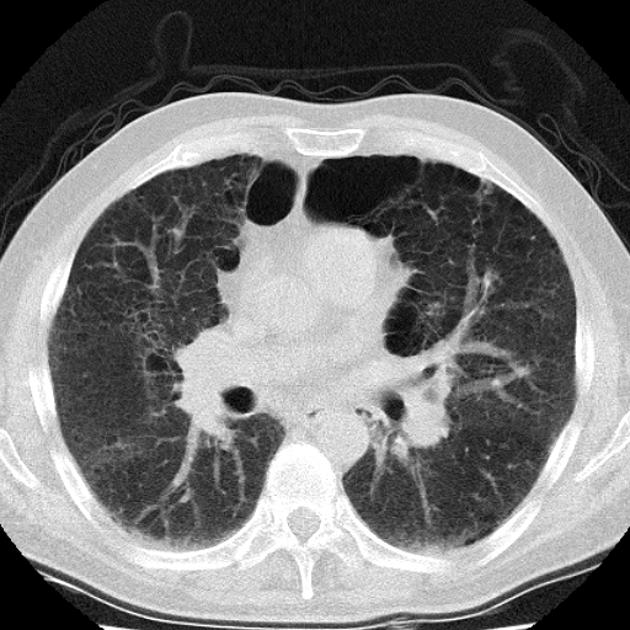
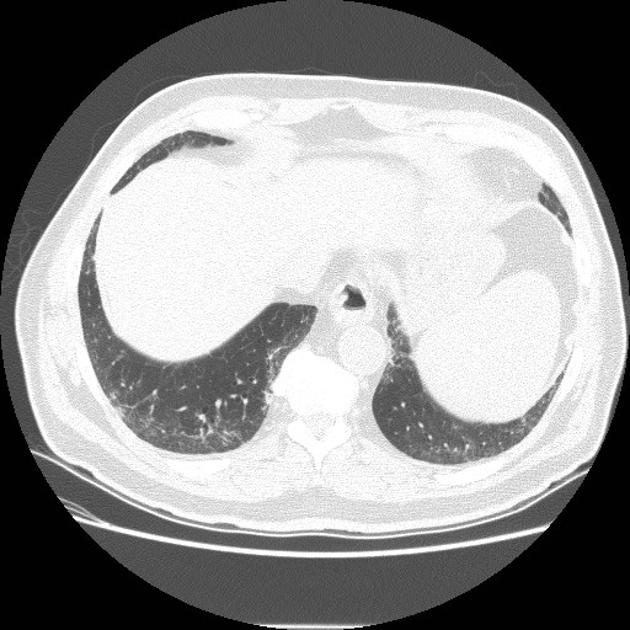
honeycombing: particularly if it involves more than 5% of the lung parenchyma, is highly specific for UIP. In general, UIP can be divided into two groups, those with <5% honeycombing and those with >5% honeycombing. It mainly reflects the stage and severity of the disease. Those with less than 5% honeycombing may pose diagnostic difficulty as differentiation from NSIP on imaging can be impossible; however, these still follow similar prognosis as other UIP patients 2
reticular opacities: in the immediate subpleural lung, often associated with honeycombing and traction bronchiectasis, with peripheral and lower lobe predominance, is considered a very good differentiating feature from patients with NSIP and concurrent emphysema 2
-
reticular opacity-to-ground glass opacity ratio: one or greater
ground-glass opacities: usually less extensive than the reticular pattern and almost never seen in isolation - usually happens in areas of reticulation or honeycombing
lung architectural distortion: which reflects lung fibrosis and is often prominent
lobar volume loss (predominantly lower lobes) is seen in cases of more advanced fibrosis
Some authors have proposed signs within a UIP pattern that are more suggestive of a connective tissue disorder interstitial lung disease over idiopathic pulmonary fibrosis, including 22:
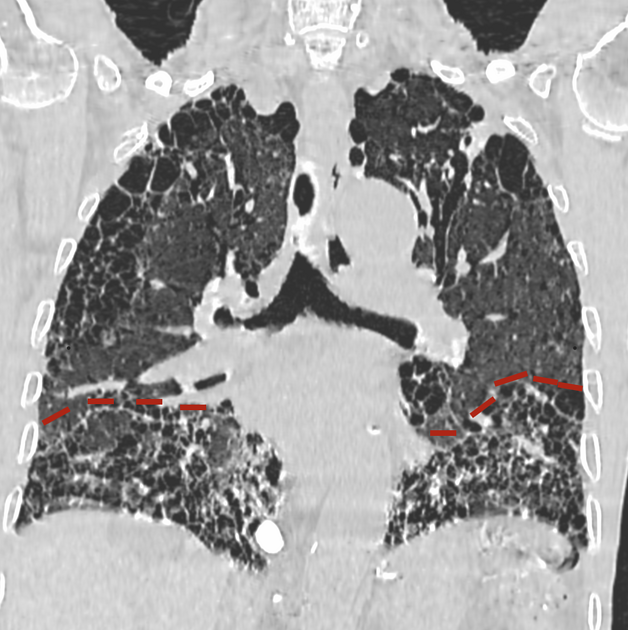
straight-edge sign: fibrosis isolated to the lung bases with a sharp demarcation best seen on coronal images in the craniocaudal plane between fibrotic lung inferiorly and spared lung superiorly, without significant extension along the lateral margins of the lungs
anterior upper lobe sign: fibrosis concentrated along the anterior aspect of the upper lobes with concomitant lower lobe involvement but relative sparing of the other aspects of the upper lobes
exuberant honeycombing sign: prominent honeycomb-like cyst formation occupying more than 70% of the areas of the lung affected by fibrosis
In some instances a classical propeller blade distribution may be seen 24.
Diagnostic criteria
Two leading societies have published criteria for the diagnosis of UIP based on HRCT findings:
diagnostic HRCT criteria for UIP pattern - ATS/ERS/JRS/ALAT (2018)
diagnostic HRCT criteria for UIP pattern - Fleischner society guideline (2018)
Treatment and prognosis
In patients with UIP, areas of ground-glass attenuation tend to increase in extent or progress to fibrosis despite treatment 8,13. In those with more active inflammation involving the pulmonary interstitium, there is a faster progression of honeycombing in long-term follow-up 10. The average rate of progression of honeycombing in patients with idiopathic usual interstitial pneumonia according to one study was 0.4% of lung volume per month 7.
Differential diagnosis
A key imaging differential on cross-sectional imaging would be:
-
fibrotic hypersensitivity pneumonitis
<25% of cases may closely mimic UIP pattern in distribution
upper zone centrilobular nodules and areas of air trapping are useful pointers to the correct diagnosis, if present
amiodarone lung fibrosis: helpful clues are the presence of hyperdense pulmonary nodules or hyperdense liver on a non-contrast CT
systemic sclerosis: presence of patulous esophagus and correlation with hand radiographs if available can be helpful
asbestosis: bilateral pleural plaques with or without calcification
combined pulmonary fibrosis and emphysema (CPFE), especially if there is upper lobe-predominant emphysema






 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.