
Interstitial lung disease (ILD) is an umbrella term that encompasses a large number of disorders that are characterized by diffuse cellular infiltrates in a periacinar location. The spectrum of conditions included is broad, ranging from occasional self-limited inflammatory processes to severe debilitating fibrosis of the lungs.
On this page:
Terminology
Interstitial lung disease is considered a misnomer by some, as many of the diseases also involve the alveolar spaces.
Diagnosis
This is the provenance of multi-disciplinary teams; clinical, functional laboratory and CT findings are integrated to reach a consensus diagnosis, if possible. Biopsy is only helpful if it might influence treatment decisions and outcome. Surgical lung biopsy carries significant morbidity and mortality; cryobiopsy is available in some centers.
Clinical presentation
Interstitial lung diseases classically produce the "3Cs": cough, clubbing of the nails, and fine crackles on auscultation 6.
Functional respiratory tests commonly show an abnormal restrictive pattern and reduced diffusing capacity.
Pathology
Etiology
The radiological appearances are not specific for the underlying cause of diffuse lung disease in many cases. It is therefore key to determine whether there is an underlying cause for the changes. A number of precipitants can cause diffuse interstitial disease such as:
smoking 1
organic dusts (causing hypersensitivity pneumonitis)
inorganic dusts (causing pneumoconioses)
gases or fumes
drugs
radiation
infection
Eliciting a history of underlying systemic disease is also helpful since they may involve the lungs in a diffuse and infiltrative manner. Examples include:
granulomatous diseases, e.g. sarcoidosis, Langerhans cell histiocytosis
-
neoplasia
primary, e.g. lymphoma, other lymphoproliferative diseases
secondary, e.g. pulmonary metastases, lymphangitis carcinomatosis
vasculitis
inherited diseases, e.g. neurofibromatosis
-
autoimmune and collagen vascular diseases (collagen vascular disease related interstitial pneumonitis) 3
-
immune dysregulation
miscellaneous, e.g. amyloidosis, alveolar proteinosis
Where a cause is not determined, the idiopathic interstitial pneumonia (IIP) should be considered:
usual interstitial pneumonia (UIP): idiopathic pulmonary fibrosis
-
non-UIP IIP
non-specific interstitial pneumonia (NSIP): non-smokers
cryptogenic organizing pneumonia (COP): previously termed BOOP
respiratory bronchiolitis-interstitial lung disease (RB-ILD): smokers
desquamative interstitial pneumonia (DIP): end-state of RB-ILD
-
other entities
Mnemonic: All Idiopathic Chronic Lung Diseases aRe Nonspecific
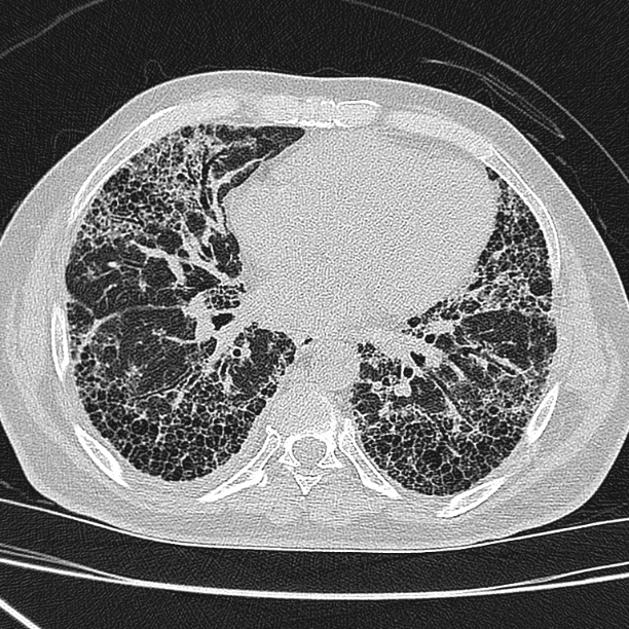
Radiographic features
The diffuse lung diseases tend to cause infiltrative opacification in the periphery of the lung, but patterns vary among the different etiologies. Please refer to the articles in each specific etiology listed above for specific details on their imaging pattern.
Follow up
Follow up and monitoring will depend on a multitude of factors such as symptoms and other co-morbidities etc. While no strict consensus recommendations are available in relation to imaging follow-up for patients with progressive fibrosing forms, many consider an HRCT at the patient’s initial presentation and then every 12–18 months to assess for progression dependent on symptoms etc 11.
Treatment and prognosis
Antifibrotics are approved for IPF and systemic sclerosis in the USA. Other fibrotic lung diseases only qualify if progression has occurred.
The extent of honeycombing and bronchiectasis, and pulmonary artery diameter appear to relate to higher mortality across all ILD subtypes whereas subpleural sparing is a favorable prognostic indicator 13.
Practical points
Interstitial lung disease is a misleading term for various diseases causing lung injury. The injury often begins within the alveoli, later spreading to the alveolar wall 12.
See also
interstitial lung pattern - radiograph



 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.