
Non-specific interstitial pneumonia (NSIP) is the second most common morphological and pathological pattern of interstitial lung diseases after UIP. NSIP is commonly associated with connective tissue disease (CTD), and a multidisciplinary team best decides the underlying diagnosis and management ref.
On this page:
Epidemiology
Non-specific interstitial pneumonia typically tends to present in middle-aged adults 40-50 years of age 1. It may be more common in the White European population 9. The overall prevalence is higher in women due to an association with collagen vascular disease, but the prevalence of idiopathic NSIP is similar in both genders.
Risk factors
Smoking is neither protective nor a risk factor for NSIP ref.
Clinical presentation
The symptoms of non-specific interstitial pneumonia include insidious onset of dyspnea and dry cough with a restrictive pattern of decreased lung function and reduced gas exchange capacity.
Pathology
Temporal and spatial homogeneity in a specimen is an essential feature. Historically, non-specific interstitial pneumonia was divided into three groups; however, due to similar outcomes, groups II and III (mixed cellular and fibrotic and mostly fibrotic, respectively) are now both classified as fibrotic type:
-
fibrotic non-specific interstitial pneumonia
more common
interstitial thickening is due to uniform dense or loose fibrosis and mild chronic inflammation
despite fibrotic changes, lung structures are still preserved
-
cellular non-specific interstitial pneumonia
less common
interstitial thickening is mainly due to infiltration of inflammatory cells and type II pneumocyte hyperplasia
lung architecture is preserved 8
better response to treatment and better prognosis
Important negative histological findings are the absence of acute lung injury, including hyaline membranes, granulomas, organisms or viral inclusions, dominant airways disease or organizing pneumonia, eosinophils and coarse fibrosis.
Etiology
NSIP is associated with a number of conditions. If there is no underlying cause, it is called idiopathic NSIP, which is considered a distinct entity ref. A non-exhaustive list includes:
-
connective tissue disorders
-
other autoimmune diseases
-
chemotherapy agents 4
thalidomide 16
slowly healing diffuse alveolar damage (DAD)
relapsing organizing pneumonia
occupational exposure
immunodeficiency (mainly HIV infection) 13
immunoglobulin G4 (IgG4)-related sclerosing disease, with or without overlapping features with Rosai-Dorfman disease 13
multicentric Castleman disease 13
Radiographic features
Plain radiograph
A chest radiograph can be normal in the early stages. There may be ill-defined or ground-glass opacities with lower lobe distribution or consolidation in a patchy, reticulonodular or mixed pattern. A bilateral pulmonary infiltrative pattern with volume loss of lower lobes may be seen in those with advanced disease.
CT
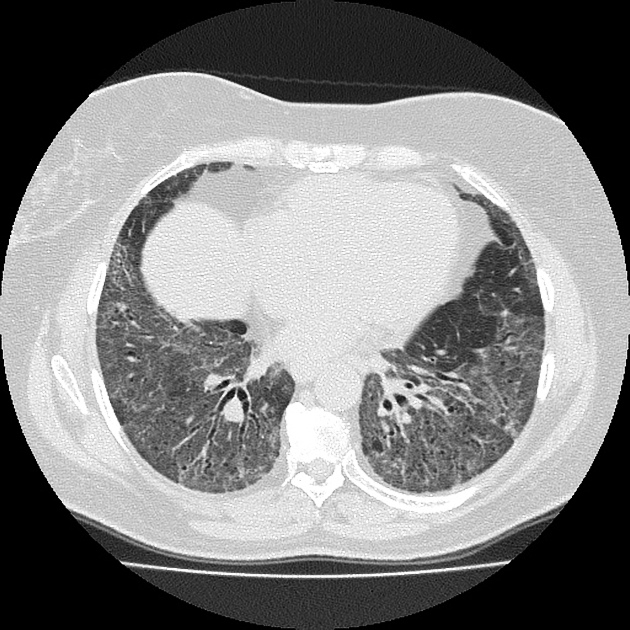
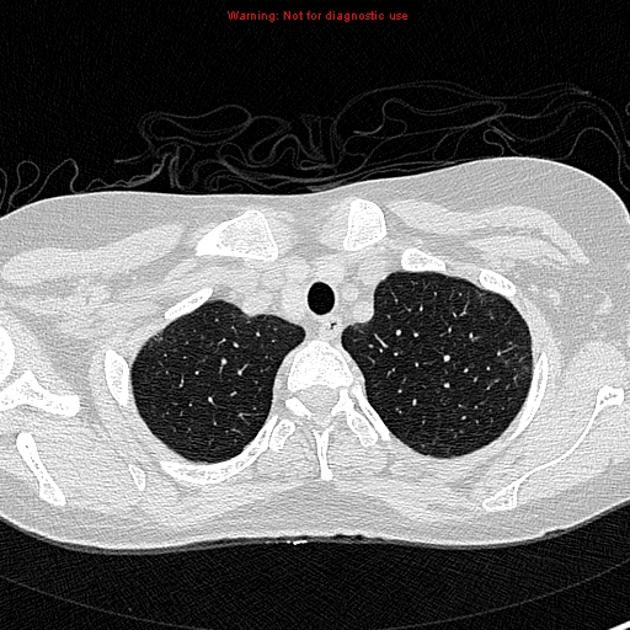
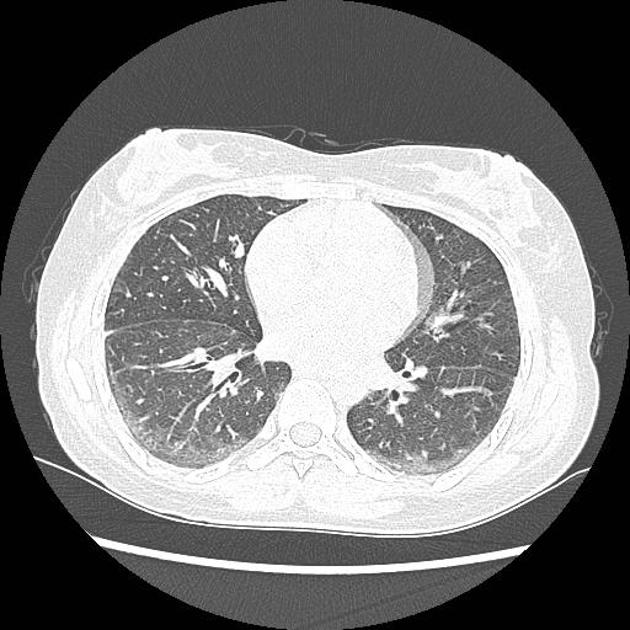
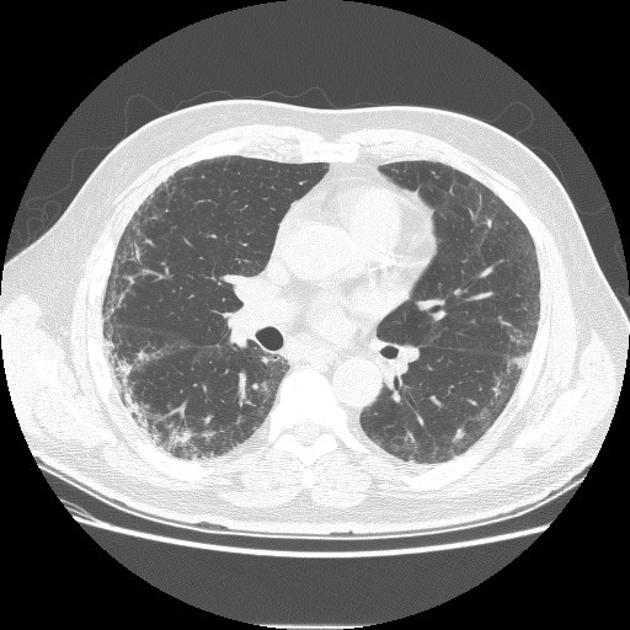
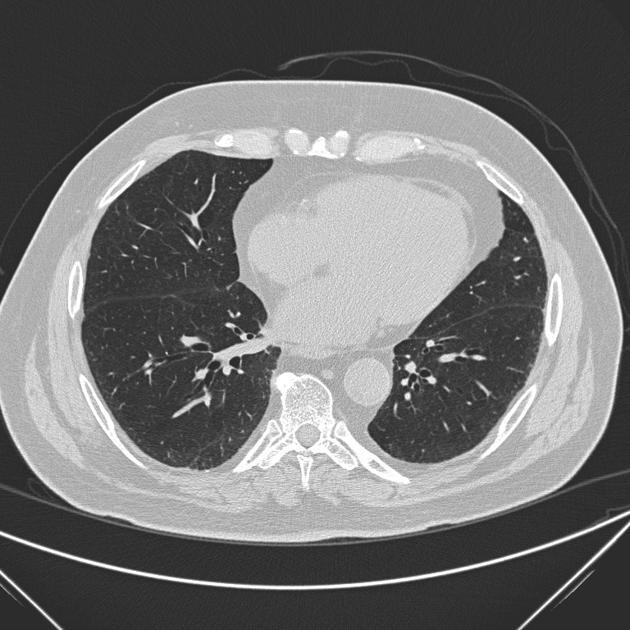
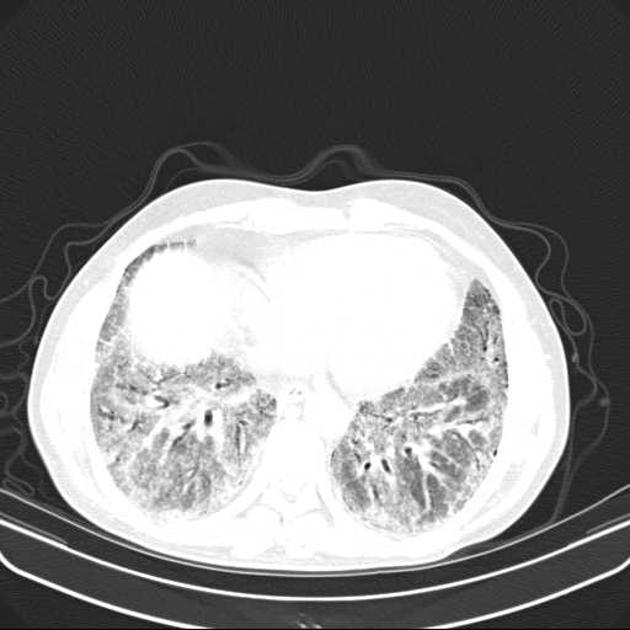
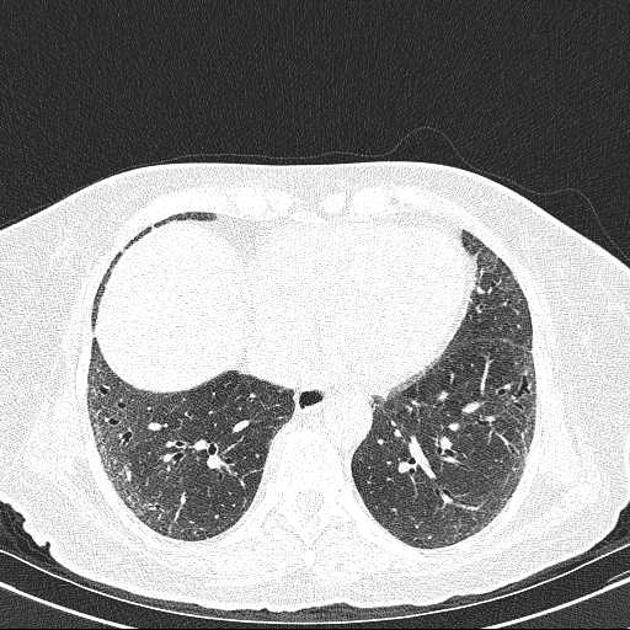
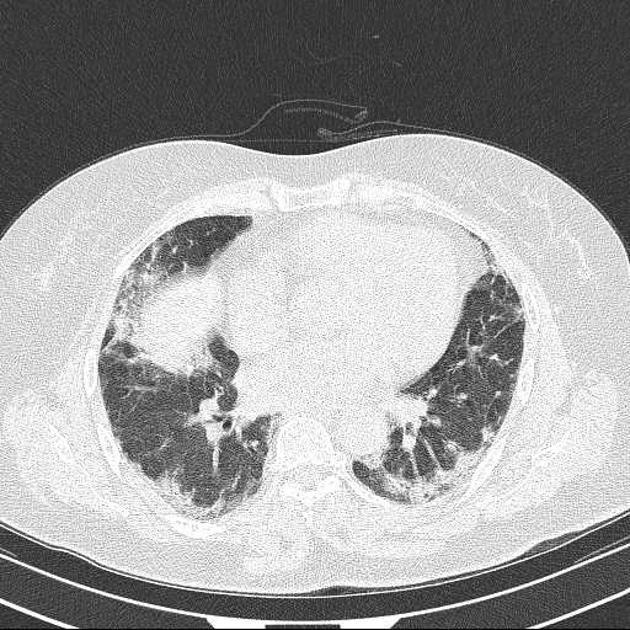
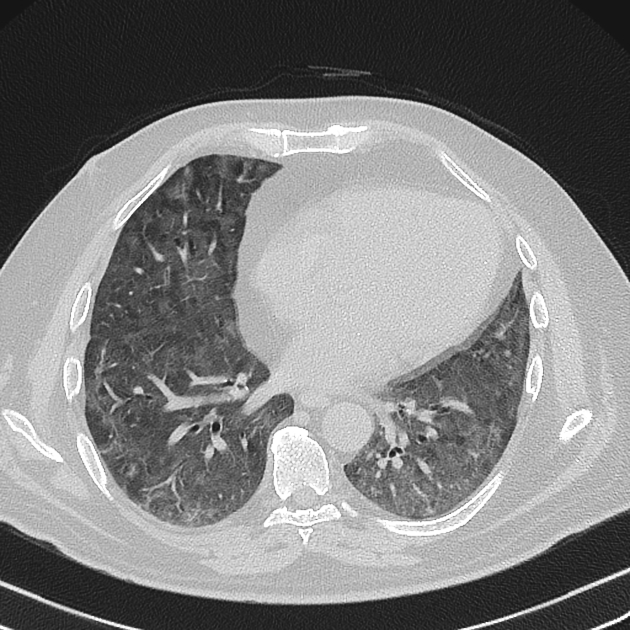
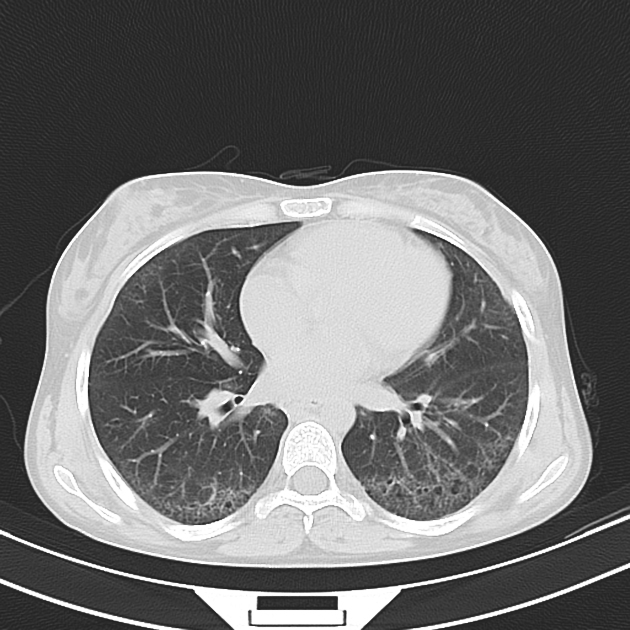
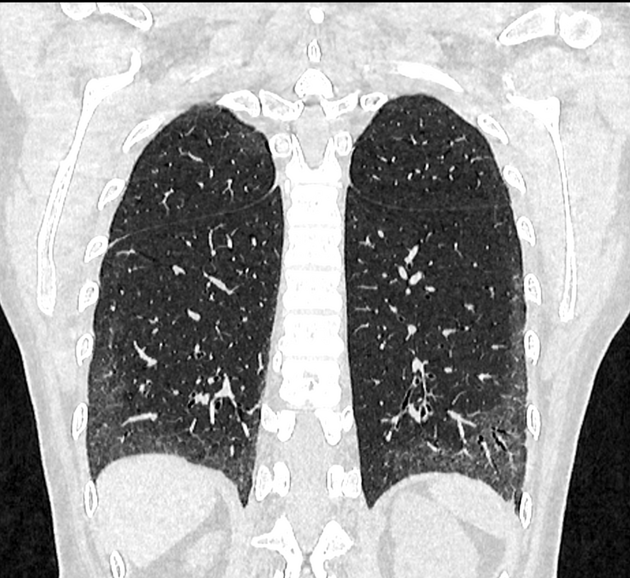
Fibrotic NSIP maximally and symmetrically affects the peribronchovascular interstitium of both lower lobes causing irregular bronchiectasis extending towards the hilum accompanied by lower lobe contraction. Relative subpleural sparing is a useful distinguishing feature when present. Ground-glass opacity may indicate cellular NSIP or fine fibrosis beyond the resolution of CT. Ground-glass opacity may also coexist with reticulation 18.
The NSIP pattern often evolves into a UIP pattern over time. Evaluation of prior CT scans is essential to correct categorization 18.
Common manifestations include:
-
tends to be a dominant feature: can be symmetrically or diffusely distributed in all zones or display a basal predominance
immediate subpleural sparing 11 is a relatively specific sign
mostly bilateral and symmetrical (~86% 14) but can be bilateral and asymmetrical (10%) or, rarely, unilateral (3%)
mostly peripheral in distribution (~68%) but can be random (21%), diffuse (8%) or, rarely, central in distribution (3%) 14
reticular opacities and irregular linear opacities (sometimes, minor subpleural reticulation), mainly in fibrotic NSIP 6,7
thickening of bronchovascular bundles: in fibrotic NSIP 6
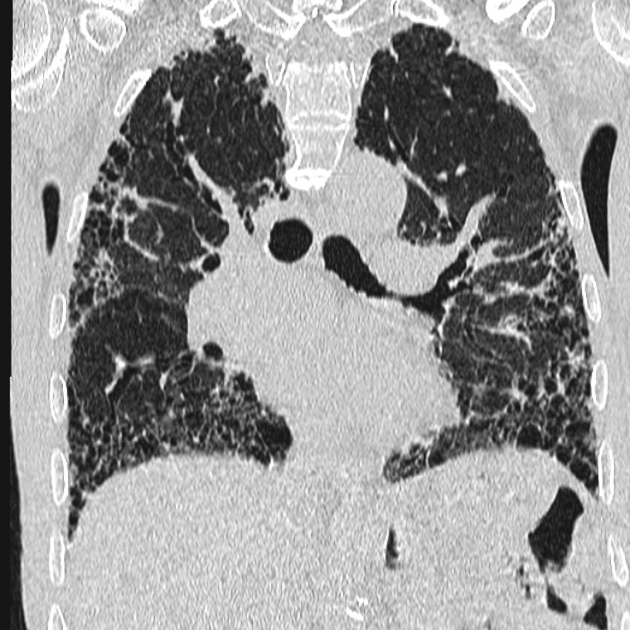
traction bronchiectasis: associated with fibrotic NSIP
lung volume loss: particularly lower lobes with traction bronchiectasis extending from the periphery to the hilum
-
in advanced disease:
microcystic honeycombing: a relatively less common feature
The presence of the following features, although they can be seen in NSIP, should make one think about other differentials:
Treatment and prognosis
In general, non-specific interstitial pneumonia (NSIP) carries a much more favorable prognosis than a usual interstitial pneumonia (UIP) pattern, with a 90% 5-year survival rate for the cellular subtype and a ~60% (range 45-90%) 5-year survival for the fibrotic subtype. Cellular NSIP shows a better response to corticosteroids and carries a substantially better prognosis than the fibrotic type. Correct and early diagnosis has a significant impact on patient outcomes because NSIP usually responds well to corticosteroid therapy or cessation of inciting causes, e.g. drugs or organic allergens 12. Mycophenolate mofetil has also been shown to improve lung function 15.
History and etymology
It is thought to have been initially described by Katzenstein and Fiorelli in 1994 14.
Differential diagnosis
The key differential is the usual interstitial pneumonia (UIP) pattern, with which there can be some overlap in imaging features 3. The features that favor the diagnosis of NSIP over UIP are symmetrical bilateral ground-glass opacities with fine reticulations and sparing of the immediate subpleural space. The presence of macrocystic honeycombing is practically diagnostic for UIP.
Practical points
Examination of prior scans is essential to correct categorization of the CT pattern.
CTD-ILD may display features which suggest the diagnosis including esophageal dilatation, serositis, joint erosions or muscle atrophy.


 Unable to process the form. Check for errors and try again.
Unable to process the form. Check for errors and try again.